Pathways and Barriers to Genetic Testing and Screening: Molecular Genetics Meets the “High-Risk Family”
FINAL REPORT
Pathways and Barriers to Genetic Testing and Screening:
Molecular Genetics Meets the “High-Risk Family”
Troy Duster, Principal Investigator
Diane Beeson, Project Director
Institute for the Study of Social Change
University of California Berkeley
The original title of this project was “Pathways to Genetic Screening: Patient Knowledges, Patient Practices”.
This work was supported by the Director, Office of Energy Research, Office of Health and Environmental Research of the United States Department of Energy under contract DE-FG03-92ER61393. The research team included Robert Yamashita, David Minkus, Duana Fullwiley, Arona Ragins, Teresa Doksum, Nadine Gartrell, and others.
To obtain a copy of this report, contact Janice Tanigawa; Institute for the Study of Social Change; 2420 Bowditch Street; University of California; Berkeley, CA 94720 (510/642-0813, Fax: -8674).
Introduction
The goal of this research has been to help map and better understand the processes by which genetic knowledge is integrated into the lived experience of “high-risk” family members. This study was designed to clarify the social and cultural dimensions of what is likely to be the proliferation of genetic testing and screening. Our strategy was to focus on both the barriers and bridges to the use of genetic information by moving beyond the medical center or clinic, into the natural settings (Duster, Matza, and Wellman, 1979). We wanted to learn what it means for families to have these new reproductive options and we wanted to examine the consequences for the quality of family life.
[footnote: Genetic screening is distinguished from testing primarily by its target population. Testing is the use of specific assays to determine the genetic status of individuals already suspected to be at high risk, while screening focuses on the general population, or at least a larger population thought to be at risk-in contrast to individuals (OTA, 1992:4). While our immediate focus in this study was on individuals considered “at high risk,” those with a family history, the larger purpose of the study was to learn from high-risk families in the hope that this knowledge would be useful to the public as a whole as genetic diagnostics become more widely available.]
The mapping and sequencing of the human genome has been described as the first major step to a new way of thinking about, and addressing, human health and illness. The Human Genome Project is often characterized as a turning point, unlocking mysteries of human nature and developing the foundation for the elimination of many forms of disease (Kevles and Hood, 1992; Bishop and Waldholz, 1990; Kitcher, 1996, Nelkin & Lindee,1995). Analysts have noted that molecular genetics, as well as the many related new technologies and analytical tools, are having an impact far beyond the realm of human health as well. For example, social institutions such as families, education and business have experienced an array of new challenges and opportunities as a consequence of discoveries in genetics (Powell, 1997; Fujimura, 1997).
Much has been written about the ethics and potential social implications of the rapidly proliferating new technologies. Concerns are reflected in the form of a growing body of legislation and the establishment of a number of new oversight and policy developments at both the state and the federal level (New York Times, Oct. 18,1997; Rothenberg, 1997). These bodies have relied heavily on the expertise clinical geneticists, legal and social expertise of the Ethical, Legal and Social Issues Program of the Human Genome Project.
While the current perspectives of scientists, educators, legislators and business people are important, the legitimacy and ultimately the public commitment to genetic research and technology will also turn in large measure upon the experience, perceptions, and expressed views of those whose lives are touched by the new discoveries and technologies. The public will certainly approve interventions that promise good health, or at least help to eliminate clear threats to health. However, the responses to genetic interventions will be influenced by how we interpret risks and costs and how we balance these options. This study explores these issues in relation to some of the most widely implemented and rapidly proliferating interventions arising out of molecular genetics — genetic testing.
The term “genetic testing” refers primarily to the practice of obtaining information on an individual — oneself or even potential offspring — by analyzing DNA. Genetic testing may be used predictively or diagnostically. When populations are targeted, such testing is referred to as “genetic screening.” These processes can identify a rapidly growing number of genetic conditions or alleles for such conditions. However, even the most optimistic acknowledge that there currently exists, and may be for some time, a long time gap between capacity to diagnose and any possible therapeutic intervention.
We are currently in a period in which genetic tests can predict with varying degrees of accuracy the risk for potentially lethal or debilitating genetic conditions from Tay Sachs and Huntington’s disease to breast cancer and Alzheimer’s disease. Yet the therapeutic interventions such a diagnosis permits are quite limited. Furthermore, diagnostic tests are proliferating far more rapidly than effective treatments. In this period of waiting for an effective intervention strategy, which could take many years, a wide range of social, ethical, economic and legal issues surface for medical and health practitioners and couples at risk for having a child with a gene disorder.
These issues are being debated from the perspective of a variety of experts, but far less attention has been given to the perspective of the families at risk who must negotiate this increasingly difficult terrain of both new and yet often still quite limited possibilities. This project was conceived out of concern for the consequences of these developments for families living with genetic disease, and/or at risk of genetic disorders. We wished to provide a forum and even a megaphone, for the expression of these experiences and viewpoints.
Often, current research on responses to new biomedical technologies is conducted in medical centers. Participants in such studies tend to be those who have already made the decision to seek medical intervention of one form or another. Patients in these settings are likely to have had multiple opportunities to interact with health care providers. Medical-center patients learn to interact in a way that they view as appropriate to the world of biomedicine. Their behavior and expressed views in this setting are far less likely to accurately reflect the perspective they employ during their everyday lives.
We have chosen to focus on sickle cell disease and cystic fibrosis, recessive genetic conditions with different clinical manifestations that affect different parts of the population. We believe that this strategy gives us a window onto newly emerging social processes and cultural issues that extend far beyond particular conditions and their uniqueness. They help us to see how members of families integrate new genetic knowledge into personal identities, mate selection, reproductive planning and other aspects of private life. In this sense, the study’s participants and the processes they reveal, are likely harbingers of things to come for the entire population. More and more people will come to have the kinds of experiences we will describe as scientists decode the genetic basis for an increasing number of conditions.
The discovery of the gene for cystic fibrosis in 1989 provided a unique opportunity for social research. Its discovery had long been anticipated since cystic fibrosis is the most common genetic disorder among Americans of northern European descent. Cystic fibrosis, however, was not the first serious disorder whose genetic basis scientists revealed. Carrier testing for sickle cell disease goes back several decades (Bowman,1977; Duster, 1990). These two conditions are the most common potentially lethal genetic disorders within their respective populations. They not only have the same recessive patterns of inheritance but also raise similarly serious biomedical challenges and issues of information management. At the same time, each primarily affects different racial and ethno-cultural group. This permitted us to observe and analyze a naturally occurring “experiment” in the variable penetration and meaning of genetic medicine in two diverse populations.
As this report will show, we have observed a wide range of responses to genetic testing, from resistance or avoidance to vigilant utilization. Furthermore, these responses are clearly patterned along a number of social, cultural and economic dimensions. Among families at risk cystic fibrosis compared with those at risk for sickle cell disease, there are significant differences as well as powerful similarities. The major pattern we have observed, however, transcends ethnicity or genetic condition and is counter-intuitive. We found that the closer people are to someone with genetic disease, the more problematic and usually unacceptable genetic testing is as a strategy for dealing with the issue. High-risk family members who support genetic research do so primarily because they view it as leading to better care and ultimately a cure, not because they support genetic testing per se.
Although more interest has been exhibited in understanding why certain ethnic groups appear more resistant to genetic testing than others, we believe the most important finding of our study is the similar difficulties virtually all high-risk families have in integrating the discourses of molecular genetics with the divergent values and priorities of family life. In this report we will explain and elaborate on this finding.
Research Strategies
The Need for Combined Methodologies. Since the purpose of this project was to reveal cultural and social-structural variations in perspectives on genetic testing, it was necessary to use methods that would reveal as much as possible about the “natural attitude” of the subjects. In other words, we wanted to discover and document aspects of their particular social worlds, much of which we could not anticipate. Such an approach demanded that we not impose academic or biomedical vocabularies or vocabularies of motive upon our respondents that they did not bring to the setting. Instead we wanted to use procedures that would give us optimal exposure to the cognitive and emotional processes, cultural patterns, beliefs and practices related to our respondents’ own life experiences. It became clear from our pilot interviews that if we wanted people to be open with us it was important that we avoid giving the impression that we were testing knowledge. Research methods that explicitly test knowledge, are often useful and appropriate. Moreover, the results of such investigations are certainly more easily analyzed. Respondents attempt to provide the “right” answers. However, during such a process, they are less likely to reveal their concerns, feelings or doubts. We elected a combined approach of fieldwork (both observation and participant-observation), semi-structured open-ended interviews with individuals, and group interviews.
Fieldwork. Although our goal was to reach beyond clinical settings, we began with several months of extensive field work in hospitals and clinics. These included attending medical rounds and conferences at two hospitals with large sickle cell clinics. We attended cystic fibrosis rounds and conferences for clinicians at a third hospital. Occasionally researchers sat in on patient counseling sessions. In all of these settings we observed formal and informal social interaction among health care providers, patients and family members, and took extensive field notes. These activities enabled us to become knowledgeable about the workings of the clinics, increasing our understanding of the diseases and their treatments, provider-patient interactions and family relationships and responses to all of these. It also created allies and informants for us among clinicians, and taught us what kinds of situations and attitudes we should minimize or avoid.
A second focus of our field work was non-medical settings. Members of our staff observed and participated in social activities and benefits for both sickle cell and cystic fibrosis patients. Such events included support group meetings, lay conferences, picnics, talent shows, sporting events and children’s camps. This wide ranging exposure to the social worlds of patients and their families enabled us to gain a more highly textured picture of the concerns of patients and their families. It also prepared us to recruit and interview in a more sensitive manner.
Once our interviewers had completed the requisite weeks of field work, they had also become acquainted with health care providers and members of families we were targeting for our study— those in whom at least one member was known to have sickle cell or cystic fibrosis. Nine of our staff members participated in local, national and international conferences or meetings of geneticists, served on study sections for genetics-related projects or served on panels and committees. While some of these meetings focused on highly technical issues in molecular genetics, some focused on the intersections between science and medicine or basic and applied science. These meetings gave us a deeper understanding of the developments in genetics and of the differences in perspectives among scientists, clinicians and patients and their families.
Recruitment and Sampling. Our first formal interviews were with mothers of young children with sickle cell disease whom we met in clinical settings. Our sampling procedures combined opportunity sampling to identify families; cascade sampling to move through the family system, and theoretical sampling as we made efforts to include differing types of families with wide ranging experiences in terms of income levels, education, religion, severity of and proximity to the disease, and age of the affected person. Our sample, therefore, while purposive rather than random, enabled us to identify recurrent patterns and ranges of responses to genetic testing across a wide band of social life. Where family members were located within traveling distance (usually about 50 miles) and welcomed us into their homes, interviews were conducted in person. Some interviews were also conducted in community sites, community centers, churches, etc. When necessary, often because of distance, we conducted interviews by telephone (34% were telephone interviews).
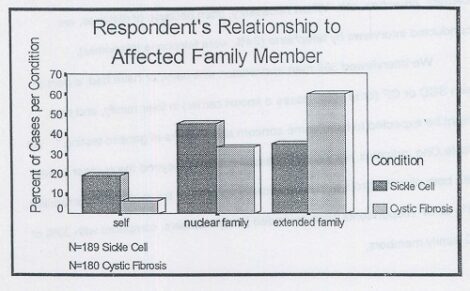
We interviewed 369 men and women who have, or have had, a person with SCD or CF (or in some cases a known carrier) in their family, and thus might be expected to have some concern about issues of genetic testing. As Table One indicates, we were successful in moving beyond the nuclear family with both disease groups, but significantly more so in the cystic fibrosis families; 61% of CF respondents were extended family members, compared with 33% of SC family members.
[footnote: Our first year of funded fieldwork and interviewing ended May 31,1993. During that year we prepared much of the groundwork for recruitment of respondents and conducted intensive interviews with 165 individuals in 40 families (See progress report #1). From June 1, 1993 to March 4, 1994, we were forced to cut our operations to a minimum due to a funding hiatus. However, the assistance of a supplementary grant from DOE during that period enabled us to keep the project going with only partial loss of trained staff. Based on a progress report renewal application funding was then resumed at a level that permitted us to continue data collection and move into data analysis. This second funding period ended October 31, 1995. We were granted a one-year no-cost extension to October 31, 1995 and again in 1996 to give us more time for cataloguing and coding data for analysis.]
Table One
Although several factors help explain this difference in levels of response, the one most often articulated was the greater mistrust among African Americans of university, and biomedical researchers. Before considering this issue further, we need to explain our methods in more detail.
[footnote: We tried to mitigate this with a general practice of matching interviewers and interviewees by ethnicity and race]
There is a two-pronged common misconception about “qualitative methodology” that should be addressed and countered, which the research reported here confronts directly. The first misconception is that qualitative methodology is either preliminary or oppositional to quantitative analysis. The second is that social behavior is so randomly distributed that a random sample is either the best or the only way to obtain compelling evidence for apprehending patterns of behavior. Both misconceptions are handled by the idea of “the saturation of the categories.”
After reviewing the results of interviews with hundreds or even only scores of different subjects, patterns always emerge in data collection; interviewers begin to report that they have heard many other respondents give similar accounts. When analyzing the data, the categories that emerge and become the basis for sorting responses start to fill up. With more and more data collected, a routine phenomenon emerges, “the saturation of the categories” (Glaser and Straus, 1967:107). This is actually a quasi-quantitative rendering of what happens in “qualitative” research.
[footnote: Psychiatrists who have practiced for several years are aware of this phenomenon, since they report such things as “when it come down to it, there are only five stories with variations around key themes.”]
The effective use of the saturation of the categories is assured not only by examination of a number of cases, but through a procedure known as “theoretical sampling.” As generalized relations among categories become discernible, hypotheses are formulated and tested by actively looking for cases that do not necessarily fit the emerging pattern. This strategy led us to search out the widest possible range of respondents along a variety of dimensions. By asking our interviewees, various informants, a member of the federal judiciary, university students and custodial workers, we located families whose members came from of a wide range of religious persuasions, economic statuses, and occupations. Our priorities also included being alert to stories of individuals and families with a range of values and experiences vis-à-vis genetic disease and technologies. For example, in spite of the high correlation between these two conditions (CF and SC) and ethnicity, our sample includes 11 African-Americans from families with cystic fibrosis and two whites and two Mexican-Americans in the sickle cell group.
The process of achieving category saturation during the sampling process clarifies some otherwise less notable dimensions of the social world. For example, we noted a number of barriers to participation in our research among African-American families, including a markedly lower level of economic resources, which in turn reduced this population’s interest in and willingness to participate in research. Enlisting their cooperation was much more difficult. However, we were usually able to overcome these obstacles using a variety of strategies. The first was to institute a policy of paying respondents twenty dollars for their participation. We offered this to all respondents. This not only made the potential interviewees more receptive, particularly the poorest ones, but “entitled” our interviewers, who before being authorized to offer this incentive, often reported being inhibited by a feeling that we on the project were exploiting busy, beleaguered family members.
A second important strategy in the success of our overall recruitment efforts was the matching of interviewees and respondents by race/ethnicity. For example, we found that interviewers of African-American descent were much more successful in gaining the trust and thus participation of African Americans and ultimately far more successful in penetrating to other family members than were our white interviewers. White interviewers were rarely able to interview more than one or two members of the same family even with the twenty-dollar incentive. We also found that the interviews conducted of African Americans by African Americans contained much more candid and critical perspectives on a number of central issues than those white interviewers conducted. The ability of the respondents to identify with interviewers by ethnicity would become an issue later, as well, when we began interviewing Asian-American families.
Interviews. We developed an interview guide to assist our interviewers, who consisted of students (mostly graduate) and senior staff members, in eliciting narratives related to six general topic areas. Within these six areas, we developed a series of open-ended probes that explored the specific terrain relevant to each interviewee:
I. History of personal experience with CF/SC
Here the interviewer would begin by asking how the respondent first learned about CF or SC and follow through their entire personal history probing for appropriate details, including thoughts and feelings and behavior in response to specific events that the respondent described as significant. This section was designed to lead into or overlap with other topics.
II. Perspectives on the disease
Here our intent was to explore the respondent’s beliefs about the meaning of the disorder in the lives of those affected. We wanted to know what they regarded as the best response to the threat of the disease, including their perspectives on prevention and treatment.
III. Genetic testing
In this section we hoped to determine whether they were aware that testing was available and how they felt about carrier testing and prenatal diagnosis.
IV. Family communication
We also wanted to explore family members’ responses to CF/SC and carrier testing. We wondered how they discuss it, how it comes up and what is said. We wanted to know about siblings, parents, grandparents, aunts and uncles, and about differences in responses between men and women.
V. Communication with friends and acquaintances
Here we were interested in whether they talk with people outside of the family about CF/SC, under what conditions, and how they perceive the responses of others.
VI. Health care
We asked what their and their family’s main concern is related to health care. We asked what, if anything, they would change about their health care and whether they have any concerns related to health insurance or coverage.
It became clear early in the process of interviewing that to the extent interviewers were intent on getting answers to every possible question, the interviewers would often derail the respondent from candid responses, or from revealing their own priorities. Therefore, we made a strategic decision to sacrifice efficiency and consistency of form for validity. That is, we attempted to allow maximum latitude for respondents to frame the issues and express them in their own way. We encouraged them to talk about those issues that were most salient for them rather than spending significant amounts of time on issues that they may not have considered. This decision arose partly from the experience of participating in previous research projects dealing with sensitive subject matter, in which it is only after the official interview is over and the tape recorders are turned off that respondents often begin to tell researchers what is really on their minds.
We were encouraged in our efforts toward a more informal and flexible approach by some of our least formally educated African-American respondents who indicated, sometimes more directly than many other respondents, that they would tell us only what they wanted to tell us. This was best exemplified in the frank formulation of a 63 year-old grandmother of a child with sickle cell disease. After initially hesitating to consent to an interview, she said: “Okay, come right now, and don’t ask me any questions, just let me tell you what I know.”
The more personal style of interviewing we adopted, while more time consuming and difficult to analyze, often results in more candid and honest responses as the interview takes the form of a supportive and very personal conversation. Even with an informal and open-ended interviewing style, the interviewer always faced a challenge in gaining the trust of the respondent — shifting the context from a formal, technical and impersonal exchange to a more intimate one. When confronted with the subject of genetic disease, particularly by university researchers, respondents often begin by engaging in a somewhat formal and impersonal discourse.
We developed a number of strategies to encourage respondents to go beyond this “official presentation of self” and to reveal the experience of their private worlds. These efforts have paid off by giving us personal accounts that are often more emotionally charged and inconsistent with the front-stage public exchanges. It is from this interplay of front and backstage discourses of family members that the current analysis emerges.
Focus groups. We began conducting focus groups in the third year of the study. Group interviews can serve a variety of purposes. The focus group interview is a well-established technique for identifying the range and patterns of opinions, attitudes, values and feelings on specific topics (Bellinger, Bernhardt and Goldstucker, 1976; Hedges, 1975; Merton, Fiske & Kendall, 1956; Morgan, 1988; Morgan and Spanish, 1984), but our intent was somewhat different and is based on the previous work of our Principal Investigator (Duster, 1990). Our goals were to verify the authenticity and salience of themes that emerged from the individual interviews. We were well aware that the most sensitive of individual interview techniques involve assessments on the part of the interviewees as to what the interviewer’s perspective and interests are and varying degrees of editing of the respondent’s remarks to conform to those expectations. Duster’s (1990) work made it clear that we would most likely get the clearest articulations of those perspectives by creating contexts in which individuals who shared particular relationships to the conditions under study (homogeneous groups) outnumbered interviewers. In other words, their shared experience was most likely to find its purest voice in settings in which it was the dominant perspective and the audience was perceived as empathetic. It was precisely these cultural dimensions of the issues we wanted to highlight.
This aspect of the process, although cumbersome to arrange, provided some of the richest data in the study. We assembled 15 focus groups, each of which were homogeneous by race and with regard to CF or SC, and stratified around key social dimensions (primarily relationship to a person with the disease). For example, some groups consisted only of mothers of an affected person, others only fathers, or siblings, or grandparents. The groups ranged in size from 3 to 8 participants.
Table Two
Focus Groups
| No. of Groups | Total Participants | Average Size | |
|---|---|---|---|
| Cystic Fibrosis | 7 | 37 | 5.3 |
| Sickle Cell | 8 | 34 | 4.3 |
| Total | 15 | 71 | 4.7 |
Our sickle cell groups were all led by African-American staff members. The cystic fibrosis groups were led by white team members or a black and white team. We found that the supportive atmosphere of these focus groups brought forth intense and animated interaction among participants. It was quite common for respondents to linger and continue their discussion after the researchers indicated that the time planned for the group had run out.
Our goal here was somewhat different than the more common uses of focus groups in which individual responses are analyzed. Instead we focused on the emergent properties of the discussion. In this setting we found that certain themes emerged as particularly salient and emotionally charged. This occurred with a force and clarity that was often less apparent in individual interviews in which the individual lacked the support or reinforcement of others who shared the same experience. Examples of charged themes that clearly emerged in this context were: anger and frustration about problems getting insurance coverage and reimbursements (especially among CF families), mistrust of government, science and medicine (among African-American SC families), and preoccupation with treatment issues (among mothers, caretakers and affected adults of both groups).
Ethical Issues
Semi-structured interviews as a research tool pose different, and often more complex kinds of ethical issues than questionnaires or pre-coded interview schedules. The three issues that were most salient were related to recruitment of respondents, confidentiality, and misinformation on the part of respondents.
Recruitment of respondents. The first issue arose early in the study when we began attempting to recruit mothers of children with sickle cell disease from a hospital clinic. It was apparent to our interviewers that these women were having a difficult struggle to meet the demands of everyday life and the needs of their children with sickle cell as well as the needs of their other children. To ask them to participate in an interview that required a significant time commitment and perhaps even transportation and child care often seemed insensitive and unreasonable.
Our observations of the obvious inconvenience to our respondents of lengthy interviews led us to the decision noted above, to offer twenty dollars to interviewees as compensation for these impositions. This offer increased the willingness to be interviewed for many and reduced the sense of the interviewers that they were imposing on interviewees. In view of the often highly emotional nature of the interviews, we decided to pursue interviewees only gently, and to accept repeated evasions as an indication of reluctance to participate.
Confidentiality. We noted early in the study that members of a family often have markedly different interpretations of major family events and may make harsh judgments of each other. Often family members are aware of these differences. This meant that the most highly-conflicted families were the ones in which recruiting study participants was most difficult and confidentiality was a constant concern.
The issue of confidentiality was particularly sensitive in this study since we often had a broad spectrum of information about our respondents and their relationships. We often found ourselves knowing more about what relatives think about other members of their family, or the choices they have made, than some family members knew. By the time we interviewed more than one family member we often had information about feelings or events that could have been disruptive if revealed to another family member. In one family, for example, our first interviewee was a woman whose affected child with CF was not her husband’s. In another case a mother of a child with CF would not refer us to any relatives because two family members living with CF kept their diagnoses secret from their employers. One of them was keeping this information from clients because he believed this was necessary for success in his profession.
Some adults were open about their CF or sickle cell, but did not want it to become any more of an issue than necessary with other family members. We dealt with these issues by establishing a policy of never referring to the content of other interviews and constantly reminding ourselves and each other of the sensitivity of our situation. It required constant vigilance not to inadvertently divulge information one family member provided to another. We could usually get more interviews per family if the same person interviewed all of them, most assuredly in those cases where the family came to like and trust interviewers who reappeared periodically to interview others who shared a household or neighborhood. We also found that interviewers could get better information if they stayed with the same family. They could ask about issues they learned about from other interviewers without divulging the specific knowledge on which the question was based. Since many of these family stories were dramatic and painful this often exerted enormous emotional pressure on the interviewers necessitating lengthy debriefing sessions and flexibility in scheduling interviews.
Since each family is unique and many family stories that reveal tensions would be recognizable to its members if described comprehensively, and since secrets that family members keep from other family members are common, reporting on family dynamics is potentially very dangerous to family relationships. This concern for confidentiality or secrecy by respondents sometimes interrupted our cascade sampling when family members declined to refer us to others.
Sometimes when the interviewer was able to get a referral, we declined to follow up on it for ethical reasons. One such case involved a young man with a mild case of cystic fibrosis. In a delicate and emotional interview, he told the interviewer about how difficult it had been for him, after four months of a relationship, to finally reveal to his first serious girlfriend that he had cystic fibrosis. He was very relieved that she did not reject him. Interviewing her could have been very helpful in understanding issues young people face in dealing with disclosure about genetic disease in the context of negotiating intimate relationships. The interviewer cautiously explored the idea with the young man, and he was willing to ask his girlfriend if she would consent to participate in the study. However, on reflection, it was clear to the interviewer that the impact of the interview on the girlfriend’s perceptions of the significance of his condition might be greater than the young man anticipated. The interviewer felt an ethical obligation to decline to interview her and told him she decided it really wasn’t necessary, since she wasn’t really a family member. The senior staff supported this decision on ethical grounds.
Misinformation. We had to be aware that even when family members gave referrals freely, they could have consequences the family members did not anticipate. For example, occasionally we would come upon a respondent who was not aware that cystic fibrosis or sickle cell was an inherited disorder, or they had grossly incorrect information about the consequences of these conditions. Our strategy was primarily to listen. When asked for information, we reiterated that we were not physicians or genetic counselors, and we made referrals to medical or patient advocacy sources. At the same time, we felt it would be unethical to withhold basic information about these conditions that was common knowledge among the more informed, particularly when they asked us directly. For example, we would occasionally run into people who assumed that these conditions were invariably fatal at an early age. On those occasions, we would point out that recent advances had improved the prognosis and suggest a source of up-to-date information.
These were not the only ethical issues we confronted. We made decisions not to pursue an interview on occasions when potential respondents warned researchers that their neighborhoods were not safe for outsiders. These choices sometimes conflicted with the goals of the research project. At times this meant pulling interviewers out of the field because they felt emotionally overwhelmed by the suffering they were being exposed to as a result of the diseases or poverty of some of the families.
Quantitative and Demographic Description of Respondents
This project has yielded a rich mapping of the social and cultural patterns of family life as it relates to new genetic knowledge and to certain genetic disorders. Our work can be likened to the mapping of the human genome in that we have identified a number of significant features of the terrain and have laid the groundwork for continued, more detailed analysis. In order to understand these patterns, it is helpful to consider some of the demographics of our sample. Table Three indicates that the educational level of our sickle cell and cystic fibrosis respondents is comparable, with a modal educational level of “some college” for both groups. The cystic fibrosis families had somewhat more members with graduate school education and fewer who were not high school graduates.
The relatively small differences in educational level stand in marked contrast to the sharp differences in income. Rather than mirroring the similar educational patterns we find that the modal income level for the sickle cell families is under $10,000, our lowest income category, while the modal income level for the cystic fibrosis families is over $50,000, our highest income category. The contrasting patterns can be seen by comparing Tables Three and Four.
(Tables Three and Four not included in online version)
Attitudes toward participating in research differed sharply between the two groups. This was apparent in the greater difficulty we experienced in recruiting participants from sickle cell families, their greater difficulty fitting interviews into their schedules without interruption once they had agreed, and the higher levels of suspicion and cynicism about biomedical research conveyed during the interviews. All of these were factors in the greater proportion of extended family members we were able to gain access to in the cystic fibrosis as opposed to sickle cell families which Table One illustrates.
We found interesting differences in willingness to be interviewed not only by ethnic group, but by gender as well. Women were much more deeply involved in the day-to-day care of disabled children than men in either ethnic group. This was reflected in their disproportionate presence in medical clinics and advocacy organizations and greater willingness to talk with us about these matters. We were reminded in a wide variety of ways that the issue of genetic disease is a far more difficult topic for men than for women. Men seem to feel greater shame about their association with deleterious genes and greater reluctance to believe they could carry genetic disorders. When they learn they are carriers of a deleterious gene, they tend to be more reluctant to communicate that information to us or to potential partners.
African-American men were less accessible to us as potential interviewees (see Table Five) than African-American women or white men. This is partly due to the higher proportion of single mothers in this ethnic group, but it may also reflect the greater mistrust of—and alienation from—the health care system by black men. The interviews with African American men, particularly in focus groups, revealed far more mistrust and cynicism about health care, medicine and science than we found among the predominantly white men or women of either group.
Within each cultural group we found clearly discernible class and familial variations in responses to genetic testing as well as examples of unique and creative responses to the challenges that genetic disease and genetic testing presented.
Respondents’ Experience with Genetic Testing Quantified
Carrier Testing. One aspect of the respondents’ relationship to genetic testing is easily quantified — their genetic status. By this term we mean to indicate their perception of whether or not they are directly affected by deleterious genes. In the case of recessive conditions such as CF and SC this usually corresponds with whether they are homozygous for the condition, carriers or tested non-carriers. In a very few cases (6 SC and 3 CF), respondents provided contradictory or obviously incorrect information or could not remember whether or not they were carriers.
A much greater proportion of the sickle cell respondents compared with the CF respondents have undergone genetic testing and thus know their carrier status (Table Six). This difference is indicated by the fact that “unknown” is the largest category for carrier status among CF family members, and the larger numbers in both the “carrier” and “tested non-carrier” categories for SC. These proportions are consistent both with the longer history of sickle cell carrier testing and the fact that we reached more extended family members in the CF group. More of those who were tested in the sickle cell group turned out to be carriers than tested CF respondents, which we expected given the greater prevalence of the mutation at issue in that population.
(Table Six not included in online version)
We found only eight individuals who have been identified as CF carriers through genetic testing as opposed to through diagnosis of their offspring with genetic disease. We will discuss the processes that led to their being tested below.
Prenatal Diagnosis and Selective Abortion. We found fourteen women reporting the use of prenatal diagnosis in each group. This constitutes a smaller proportion of the African-American women than the white women. However, this figure tells us very little about significant differences in the reasons for undergoing such procedures or about differences in attitudes toward selective abortion.
We can see hints of these differences in the fact that it was only in the CF families, where we found two cases, that selective abortion actually occurred after undergoing prenatal diagnosis. In the first case prenatal diagnosis was offered primarily as a result of the birth of a previous child with CF. This couple had made a decision they would abort if this fetus had CF. The fetus was found to be free of CF but diagnosed with Down syndrome, and the pregnancy was terminated.
The other selective abortion for CF occurred in a family that already had two previous children with CF. The couple wanted a healthy child and planned to keep the pregnancy a secret from family members and friends until after the amniocentesis could assure them the baby would be free of CF. They chose abortion when they learned the fetus was affected. This couple later went on to have another pregnancy in which it was determined the fetus was not affected. They continued this pregnancy and now consider their family complete.
African-American family members typically disapprove of the use of selective abortion, particularly for themselves, but CF families very often reject selective abortion as an appropriate option. Twenty African-American mothers of children with sickle cell (and no fathers) reported they were aware that they carried the sickle cell gene before the birth of an affected child, yet did not intervene to prevent this from occurring. Mothers were aware of a risk in only three families that had biological children with CF. One family chose to adopt a second child with CF some time after learning their first child (who was also adopted) had the condition. These numbers tell us very little about the meaning of genetic disorders and new genetic technologies in the lives of our respondents, but they do suggest that the patterns of response among high-risk families are often not what would be expected from a purely biomedical perspective. They also provide a partial framework for understanding the meanings of genetic disorders and new genetic technologies in the lives of our respondents. We will discuss some of the reasons for such responses in the next section.
Cultural Differences and Cultural Issues: Context and Meaning
The differences between CF and SC respondents in relation to genetic testing, as the preceding tables suggest, are numerous, complex and multifaceted. Both racial-ethnic groups display a variety of characteristics and responses that reflect their differing economic positions, cultural heritages and the unique challenges of a particular disease. Nevertheless, they have much in common. Our most significant findings do not involve the pronounced differences between the perspectives of the two groups, but rather larger cultural conflicts that both cystic fibrosis and sick cell families must negotiate — the conflict between the diagnosis and communication of molecular or genetic information and the context in which that information is delivered inside families.
Diagnosis of a life-threatening genetic disorder, or the identification of a gene that could cause such a disorder, has dramatically different meanings for family members and for those who approach it from the perspective of molecular genetics. For the latter there is the analytic power of science brought to bear upon a human problem — the tracking and decoding, the probe to the molecular level that from the perspective of family life strips away other contexts — with the usually corresponding hope that this information will enhance the health and the lives of those who receive it. For the family members, however, the cold, hard, scientific facts — the “neutral” information — are never neutral because they are never without social context. The nature and character of that context varies from family to family, from genetic disease to genetic disease, by ethnic and racial group, by religious and cultural group, and by gender and social class. These contextual differences place genetic testing in somewhat different perspectives. But one thing is common to all family members who confront a genetic disorder or risk thereof — the vigorous insistence on recontextualizing what they view as a decontextualized genetic diagnosis.
Three key dimensions of the recontextualizing that individuals inevitably indicate concern with are family unity, personal identity, and social standing. These are aspects of culture that CF and SC respondents have in common that make genetic testing problematic as well as sometimes useful and empowering. After discussing these shared concerns we will examine some key differences.
Family Unity. The family is by definition a collective. When information about deleterious genes is introduced into families where one or more members is known to be disabled by the condition, the character of that collectivity is often notably altered, and sometimes vitally threatened. This is because for these families deleterious genes are interlaced with deep cultural meaning, and are never just abstract concepts. Not only are they embodied, they are unequally distributed among individuals who previously saw their family membership as something they shared equally with their kin. A previously shared legacy is suddenly redefined in a way that emphasizes a new kind of difference among family members, identifying some as having fundamental “defects”, and perhaps as having passed them on, or as potentially dangerous to their own offspring, while other members may be certified as free of such liabilities. This definition of the situation introduces classifications into the family that, however practical for appropriate medical treatment, are likely to be disruptive of previous relationships. When family members refuse to integrate genetic information into partner selection or reproductive planning, some professionals characterize this as “denial.” But this characterization obscures more than it reveals.
Historically, most health problems have been understood to be the result of threats from outside the family. Genetic disease, on the other hand, identifies a child’s parents as the source of the problem. Indeed, children sometimes refer to their condition as “a disease my parents gave me.” By identifying the source of the disease in the parents’ biological makeup, issues of guilt and blame surface. Usually these feelings are displayed inadvertently, rather than deliberately. Guilt and blame are sometimes acknowledged openly, but more often the admission of such feelings is accompanied by the claim that they were part of an adjustment process in the past, but are no longer a serious problem. A mother of a child with sickle cell was somewhat more explicit in voicing these feelings than some others when she stated:
I feel responsible and his dad feels the same way. It’s like we have done something. We have shamed ourselves real bad, but you just have to deal with it. Society puts people down about a whole lot of things. I feel they look upon me as though I’m nasty. You know, they don’t take kindly when you do something to a child.
Fathers seem to have special difficulty accepting their role in their child’s disorder. This may even lead them to question (how genuinely we do not know) whether they are in fact the biological father. A more constructive response was expressed by one father who refused to have his identity spoiled by genetic information. He insisted:
What makes me a good parent is not whether or not my child is perfect, but what I’m able to do to help her.
The recontextualization of genetic information is something that all families do. It typically becomes an occasion to re-examine some taken-for-granted assumptions about one’s identity or the meaning of relationships.
Because neither clinicians nor parents seem to anticipate the potential social significance of such information, carrier children are sometimes informed which parent’s defective gene they have received (and thus are at risk of passing on to their own children). Children with disease, who have received one mutated gene from each parent, are sometimes (and apparently this is occurring with increasing frequency) informed which parent’s cystic fibrosis or hemoglobinopathy gene scientists believe to be most lethal. The assignment of blame and culpability and the resulting guilt and shame all lurk as part of the recontextualization.
The similarities or distinctions in genotypes among children become salient as well. Parents indicate the distribution of disease and trait and this information can create divisions within the family. For example, one woman was diagnosed with CF several years before she was genotyped. One of her alleles is a Delta-F-508, the other is unknown. In referring to the second allele, she says, “I call it my good gene. It’s like the one that sort of alleviates whatever the Delta F508 does.” She never learned which parent gave her which mutation, but increasingly this information is available and must be interpreted by family members who are unable to abstractly “partial it out” from their social relations. In one family a child was told that the mutation he received from his father was the more destructive one.
The opportunities for casting blame are more obvious in the case of grandparents because they are not equally implicated in their grandchild’s condition. The neutral, technical purpose of testing them is to determine which branch of the family is at greatest risk. Nonetheless, testing invariably turns out to be a source of considerable distress for grandparents. Their first reaction to the news is usually to deny that any such condition has ever existed on their side of the family. In one case the grandmother who was tested and found not to be a carrier was sworn to secrecy by her husband. This nullified the ostensible purpose of her test, which was to learn which side of the family should be alerted. Many resist testing because they anticipate difficulty living with the knowledge that they contributed, however innocently, to their grandchild’s illness. As one grandmother asked:
Whose fault was it? Was it mine or my husband’s?…I’d like to know which one — who carries the gene; my husband or I. But on the other hand, maybe it’s good I don’t know.
Our data suggest that these feelings of blame and shame for being what several families call “the culprit” will proliferate with an increased capacity to identify a particular “imperfection” via genetic analysis. A sense of responsibility for the child’s suffering is something few parents or grandparents can avoid. Humans insist on giving meaning to new information, particularly when so much social interaction focuses on gathering it.
The issue of family unity takes on a somewhat different cast depending on some of the families’ other values. Families that are more religious and traditional have a greater level of comfort and acceptance of premarital carrier testing, but are often extremely uncomfortable with and reject prenatal diagnosis. On the other hand, couples that are more egalitarian, highly educated and not particularly religious have fewer reservations about prenatal diagnosis. Both of these types of couples may resist carrier testing before marriage because they see it as a violation of their commitment to the concept of romantic love.
Self-Identity and Partner Selection. Another key element of genetic testing is the impact it can have on the self-identity of the person affected, often related to the life stage of the individual tested. When testing occurs in infancy it becomes a pivotal focus in defining the child’s future and relationship with the parents. Testing is viewed as a powerful indicator of who the child will become and what his or her future may hold. Results of genetic tests sometimes have welcome consequences, insofar as they facilitate a correct diagnosis and thus point to treatment options that might not have been utilized otherwise. Genetic diagnosis brings relief when the parents, usually the mothers, are being blamed for their child’s problems. For example, before receiving a correct diagnosis of cystic fibrosis one mother (149) was told that the problems her infant was experiencing were due to her being an overly nervous mother, and that something was wrong with her milk supply. In this case, the genetic test provided an explanation (CF) that made her feel less guilty and incompetent as a mother, and facilitated more appropriate treatment for the malnutrition and dehydration that posed an immediate threat to her child’s life.
Genetic testing to confirm the diagnosis can be a relief to parents when it indicates to medical practitioners and other authorities that a child’s self-report of pain is due to sickle cell disease and not, for example, physical abuse, inadequate care or hypochondria, all of which are frequently suspected. It may also point to specific treatment strategies that families find more effective than those resulting from other explanations for symptoms. Thus many psycho-social benefits of genetic testing are related to diagnosis of full-blown disease.
While there is a full range of responses, the first diagnosis of sickle cell or cystic fibrosis in a family is typically received as devastating news. Family members repeatedly report being told that their child will not live to adulthood, along with other similar prognoses that have very often proven unduly pessimistic. In one family we interviewed an adult who as a child with asthma and allergies was misdiagnosed with CF. This diagnosis caused her mother to give up hope for her daughter’s future. Today, the adult daughter is very resentful that major decisions about her life were made under the erroneous assumption that she would die young:
It very much affected my relationship with her [mother]. She was always acting like I was going to die and she made decisions about my life as if I was about to die all the time.
This case is of theoretical interest because even though the diagnosis of CF eventually was proven incorrect, its social consequences remained significant. One could argue that such mistakes are much less likely where DNA testing is available. However, we have found adults with mild cases of CF or SC who were grateful they were not diagnosed sooner, because they managed to receive symptomatic treatment while living a “more normal life” and maintaining an “unspoiled” social identity.
The predictive aspect of genetic testing in the context of a struggle to make a rewarding life is rarely interpreted as enhancing one’s power. Genetic testing, as a practice, is not necessarily welcome. This is particularly true when it removes or sharply diminishes one of the main resources in care giving — hope. In response to learning about the accuracy of DNA diagnosis, a mother explained:
A mother is someone who loves the child and doesn’t want to hear all this stuff about his limitations. I don’t want to hear what’s going to happen down the road as if its all laid out, because I couldn’t take good care of him under those conditions.
The knowledge that one is a carrier of a potentially fatal genetic disorder takes on particular significance when it arises in young adulthood during or before the quest for a life partner. Medically this is one of the least controversial uses of genetic testing. Respondents may claim to favor the idea that carriers should have their partners tested. However, their actual behavior and the behavior of other family members makes it clear that there is a strong undercurrent of resistance to carrier testing. While the active and outspoken objection is apparent among a minority of respondents, more implicit indications of discomfort with genetic testing are frequently expressed. A typical response is the failure to take any action, even a telephone call, to inquire about proceeding to get tested.
One’s desirability as a potential partner is one of the most powerful issues in personal identity and biography in our culture, and the idea of romantic love is pervasive. Respondents in both sickle cell and CF samples find carrier testing a troubling, even deeply disturbing real counter to the dominant image of self and other as romantic love objects.
Evidence abounds that even when individuals have been tested and are found to be carriers (often this occurs at birth in the case of sickle cell due to newborn testing programs), this does not necessarily lead to the integration of this knowledge into partner selection or future childbearing decisions. Often those who have been tested for sickle cell trait reject the relevance and implications of testing in choosing a partner. Sometimes results are forgotten. More often carriers simply fail to communicate their carrier status to prospective mates. One young woman indicated her distaste for the idea of using carrier testing as a factor in mate selection when she compared the idea to “breeding dogs.” Another young woman explained:
Genetic problems can’t be prevented because sometimes you don’t plan a baby. They just happen. You can’t do anything about it unless you say, “Before I sleep with you I want to get you screened,” and it don’t work that way, not in reality.
Our data support a recent report of related research by Hill (1994:29-47), who also found widespread resistance to integrating sickle cell testing into mate selection. She describes this response in the African-American communities she studied as “obfuscation of SCD medical knowledge” and attributes it to SCD’s threat to motherhood and the distinctive consciousness the material realities of life engender for Black women. Our findings are consistent with hers, but we would emphasize that this attitude exists not only among African Americans, but among European Americans as well. The reasons for this resistance are deeply rooted shared values about the affective nature of close human relations as opposed to their instrumental significance.
This refusal to consider one’s partner’s carrier status is consistent with the ideology of “romantic love” — the most socially approved justification for modern marriage and for sexual activity. Respondents routinely declared that “if they truly loved someone,” they would marry them despite the fact that they both might be carriers, and that “if it’s real love, you can overcome things like that.”
For example, before the availability of carrier testing, one of our male respondents hesitated to marry a woman known to be a carrier because she had previously borne a child with cystic fibrosis. He eventually went ahead with the marriage because he decided that “marriage should be for love.” In myriad ways respondents make it clear that they view the use of carrier testing as a factor in partner selection as incompatible with what impels them to have a relationship, the expressed experience of love.
Once the crucial decision of choice of a mate has been made, genetic testing has direct relevance for reproductive planning. Most frequently this occurs around the issue of prenatal diagnosis. Highly educated, and higher income, non-religious contemporary couples are more likely and more willing than traditional, religious, working-class or lower-class couples to consider prenatal testing and selective abortion in hopes of avoiding the birth of children with serious disorders than they are to demand or expect carrier testing of prospective partners as part of mate selection. Yet, even among the most highly educated high-risk families we studied, we found significant resistance to selective abortion. Few parents in our sample expressed a willingness to use this method of preventing the birth of an affected child, and only one couple in the DF families actually did selectively abort a CF fetus. Interestingly, this resistance to selective abortion does not appear to reflect a rejection of abortion, per se. We found seven members of sickle cell families who reported they had had abortions, but none of these were related to prenatal diagnosis. Only one of these involved fear of sickle cell, but this abortion was not the result of genetic testing. Rather it was an abortion obtained by a teenager, who did not even know her carrier status. Because of her family history, she feared her child would be affected, but she did not know the carrier status of her partner.
[footnote: This is consistent with the work of Wertz, et al (1992:1077-1085), who found that only twenty percent of parents of children with CF say they would abort for CF in the first trimester; 54 percent would not have an abortion for CF.]
Respondents from both the CF and SC groups often told us that although they were politically “pro-choice,” they would not themselves abort an affected child. The experience of emotional closeness to someone with a genetic disease reduces, rather than increases, the acceptability of selective abortion. A close relationship with an affected person appears to make it more difficult to evaluate the meaning or worth of that person’s existence solely in terms of their disease. Family members consistently affirm the value of the person’s life in spite of the disorder, and see value for their family in their experiences with (and of) this member, and in meeting the challenges the disease poses. For example, a woman whose sister had died of cystic fibrosis told us:
If any one of us were pregnant and it was clear that we were carrying a child with cystic fibrosis we knew we would bring it to term. We wouldn’t have wished that Sally not be born, and the thought of not having a child because this child shared something that Sally had — it just creates such cognitive dissonance in my mind that I don’t even know what to make of it.
At another point in the interview she explains:
I understand how someone without the experience of my family could think that children need to be perfect or else they’re miserable, or we do a disservice in having them, but it’s never been part of my expectation that people are perfect, or that perfect health means perfect happiness. I’ve just never equated goodness or life meaning with perfection, and so even though I know intellectually that I should be in a better position to understand the argument that one shouldn’t be born unless one is quote unquote perfect, but it makes no sense to me.
Even when respondents indicate they are in favor of genetic testing, they often have difficulty accepting the possibility of selective abortion. In response to a question on the topic of prenatal diagnosis, one such respondent was asked, “What would your children do with this information?” She replied:
My feeling is that both of them, even if they knew they were pregnant with a child with cystic fibrosis they would go ahead and have the child. They’ve certainly seen Brian grow up and function well with the disease. They realize he is just a normal kid. He’s mentally healthy and he’s physically able to do things. I would hope that they would go ahead and have the baby.
Members of families in which there has been a diagnosis of a gene disorder make it clear that the issue of genetic testing or selective abortion cannot be considered without reference to the family member with the condition in question. The stronger the bond with the affected person, the less likely one is to favor prenatal testing and selective abortion. In some cases, highly educated individuals close to persons with CF seemed to have no trouble integrating their commitment to a person with CF and the idea of selective abortion, but we found no cases where such a decision was contemplated in practice (as opposed to hypothetically) without a high level of personal conflict.
Even the case of a couple whose first child had CF, and who subsequently chose to terminate a fetus diagnosed with CF in a second pregnancy illustrates this conflict. They were very careful to shield themselves emotionally as much as possible. They used a number of strategies for doing this. One was keeping the pregnancy a secret, particularly from other families that had children with CF. A second was by learning as little as possible about the fetus other than whether or not it was affected. The woman explained:
We chose not to find out what the sex was. I was trying not to perceive that fetus as a child. But having been a pregnancy it was hard to not think of this one as a somebody, you know with a personality.
When the abortion was done at 12 weeks she requested general anesthetic despite the fact that it was not medically necessary. Months after the birth of a subsequent healthy child, this mother still has conflicts and concerns about how to handle this information within the family. She explains these this way:
[H]ow will I- at some time I want to be able to present all this [to the children], too. How the whole context of all this affected them . . . that they might have had another sibling that we chose not to do that. Because I think that’ s important at some point. . . It’s going to be hard because I don’t know how they’ll respond.
External Forces. The third dimension of the recontextualization that individuals engage in has to do with the meaning of genetic disorders in the context of the family’s relationship with the larger society. This dimension includes the flow of economic, social and cultural resources between the family and the society. It is here that the incentives for prenatal diagnosis and selective abortion are greatest.
The issue of access to health care arose in the interviews in several ways. Sometimes respondents introduced it as they told the stories of their encounters with genetic disease, and efforts to get effective care for their family member. Other times they raised the issue in response to a question about who should have access to genetic test results. This was a question that often evoked references to insurance companies. The most typical form in which the topic was broached was under the general topic of health care, in response to the question, “What is your main concern related to health care for yourself and your family?” Because the format of the interviews varied depending on the salience of particular issues to each respondent, we will include information on context when quoting respondents.
About one-third of our respondents failed to report any concern about access to health care or insurance issues during individual interviews. Many of these respondents were healthy individuals who were not deeply involved in the care of the affected family member. A few who were parents said they felt fortunate to have excellent coverage. A parent who is also a physician employed at a large medical center explained to us that he knew he had the finest care for his child available in the world. In general, however, the more direct experience people have dealing with genetic or other disability the greater their concern, mistrust and often anger at the current health care financing system and its consequences.
(Table Seven not included in online version)
The most emotionally charged expressions of the need for health care reform come from adults with genetic disease and the parents of children with cystic fibrosis or sickle cell disease. They usually feel that their survival depends on the quality of medical care available to them, yet obtaining the care they need is often a precarious process to them, primarily for economic reasons. For example, a 29-year-old PWCF (person with cystic fibrosis) with a doctorate who is pursuing an academic career told us she worries more about insurance than about getting sick or getting better. Her biggest concern about health care is paying for her treatment. She told us she wanted to have free health care. “I want that even more than I want not to have CF.” Another young adult with CF echoed this sentiment when she told us:
If someone were to ask me which is scarier to you, dealing with CF and the thought of dying or medical coverage, without a heartbeat of a thought, medical coverage scares me to death! . . . [H]aving the CF I can cope with as long as somehow some way I’m going to be able to get the medical coverage and care that I need. . . . What’s going on with our medical coverage scares the death out of me. I have nightmares. In fact, when we got that letter about all the changes that were going on at [Smith Medical Center], I didn’t sleep, and it all hit all at once. I had stuff going on with my medical coverage, I had stuff then at [Smith], so you’re rocking my world. You’re taking away my place to get health and the way to pay for that health! That scared me way more that the fact that I was coughing up blood at the time! Way more. So those are the things that rock my world. (285)
One woman who was particularly vocal on this point was not diagnosed until she was in her twenties. When asked, “In general, what do you think it means for a child to be born with cystic fibrosis these days,” she replied:
Well, I think you have to classify the question according to the country. Because I have an international perspective, I realize that, for instance, people born with CF in most European countries are assured health care, regardless of the fact that they have a pre-existing condition. And I think that has to be stated right up front. In the majority of countries employment is assured despite having a disability or a preexisting condition. I think in the United States we are more akin to, if you will, third world countries, in our outlook as far as health care and employment availability for people with CF. I think as far as health care per se goes, you know, as far as treatment in hospitals and whatever, we are on a par with the rest of the world.
For a number of years this woman received her health care coverage through her husband’s employment. After several years of marriage she “had a really bad year.” She went on to explain:
The following year, he was laid off, and when he asked why he was laid off—a small company with 25 people, they said, “because your wife’s insurance was too high. . . . Your premiums are too high, and there was absolutely nothing we could do legally about that. . . Whenever I talk about the health care system in the United States, my blood pressure goes up.
As the life expectancy of persons with cystic fibrosis lengthen (it is now 30 in the USA, and significantly higher in some European countries, especially Denmark) new problems arise related to health insurance coverage. Most parents’ health insurance coverage for their children runs out when the children reach their early twenties. Adults with CF are often unable to be employed full time because of their physical limitations and time required for therapy. If they are healthy enough to work full time, many companies will not hire them because they fear lost time from work higher insurance premiums. As a result many adults with genetic disorders and their families feel trapped by a system in which medical and treatment advances that have enabled them to live longer are being undermined by a bureaucratic and hostile health care financing system that subverts their potential to be productive citizens. There is an elaborate discourse within the social world of cystic fibrosis about strategies for dealing with these concerns. This discourse is often infused with strong emotions, particularly fear and anger.
Early during a focus group made up of five young men with CF we asked participants about their expectations for the future. An 18-year-old with CF explained how he was planning his future around an occupation that would give him the best health insurance. In his view, this meant not being self-employed, but instead working for a large company. The focus group leader asked the next young man, “Is that an issue for you?” He replied,
Yeah, I think its the biggest issue for all of us. Lets see, I’m in the third year of a COBRA insurance policy that only lasts three years, and then the only insurance I have besides graduate student health insurance plan which is basically worthless, is the State of California, GHPP (Genetically Handicapped Person’s Program), which luckily will probably do everything I need done. But that also means I’ll probably have to either get an employer when I graduate that will somehow get around the pre-existing condition clause, or stay in California and then do something else.
Another chimed in, “We’re not in the right bracket, so its real expensive for us to have GHPP.” When a young man who had coverage was asked if insurance was an issue for him, he replied,
Not in that vein. The things that concern me about insurance are a lot more of what they do cover once they cover you. I happen to belong to an HMO that every time I tell a doctor I belong to this particular HMO they just roll their eyes and say, “Oh, they’re the worst. A typical example is — this has happened three times in the last year, where I’ll put off going to the doctor till like Friday when I gotta see somebody. I see him. Her prescribes an antibiotic for me. I go to pick it up Friday afternoon. Its late, and my insurance company has to okay this particular antibiotic in order for them to be paid. So the pharmacist has to get a hold of this insurance company to get an okay. They put him on hold, and if they don’t get it okayed I’m going through the whole weekend without it. My company had three different plans, I picked the cheapest one because the other two were very expensive. You could pick your doctor, but the cost was prohibitive.
One participant suggested stocking up on medications before they are needed. He was followed by a young man who felt his insurance company was deliberately harassing him.
I got approval for aerosols, but they won’t purchase any nebulizer units at all. Well, you can’t use this thing without the nebulizer units. And you can’t take this medicine. They’ll pay for the medicine, they’ll pay for the machine, but not the units. That’s crazy!
As the discussion revealed, this kind of situation leads to patients hoarding medications and medical supplies because they cannot trust they will be available when they need them. One young man offered the explanation that they were just trying to irritate people with chronic illness into quitting the HMO. Another offered, “It’s either get them out or kill them!”
This discussion then moved to the value of preventive treatment as opposed to acute treatment. One participant lamented the fact that sometimes choices have to be made out of fear of long-term costs reaching the policy cap. Drug co-payments were another problem. One young man mentioned that prescription costs under his policy were recently increased to eleven dollars each. He added that he requires five or six prescriptions per month. The discussion then moved to the problem of paperwork for reimbursements.
Cystic fibrosis patients and their families are very aware of the differences between health care policy in this country and Canada and other Western European countries. They are aware that differences in aggressiveness of treatment have significant consequences for their health and longevity. Denmark is occasionally offered as an example, since a Danish physician speaking at a CF Conference attended by some of our participants presented data showing the life expectancy of CF patients in Denmark is forty and rising.
Adults with CF who can avoid being officially diagnosed or labeled often do so to avoid discrimination. We interviewed a relative of two adults with CF who are hiding their diagnoses. One is in the military, and the other is a professional who seeks treatment out of state because he fears his career will be jeopardized if his condition is known. Neither of these adults would agree to be interviewed due to fears of disclosure of their conditions. In both cases employability and, more indirectly, health care coverage are concerns.
The perspective of parents on the topic of health care coverage is quite similar to that of CF patients themselves. One significant difference is that while adults with CF are locked out of jobs, their parents are often locked in. In virtually every family with a child with CF, insurance and access to good CF treatment have a profound impact on the parents’ professional and geographic mobility. In some cases women who would prefer to be full-time caretakers of their CF children must keep their jobs to keep the insurance. Fathers are often unable to move ahead professionally because they cannot risk even temporarily losing coverage.
When children grow up and are uninsurable themselves, their parents may under some policies have their child declared disabled and dependent. We have seen several parents who would like to retire, but cannot because to do so threatens to deprive their child of good health care coverage.
Extended family members often recognize the insurance issues as potentially problematic when they consider genetic testing. While the insurance situation appears to be a factor in discouraging carrier testing, our data indicate that it encourages selective abortion. For example, a cousin of a person with CF explained:
I would probably want to know whether or not I carried it…”If I was carrier, I would think very carefully about whether or not we had enough money, because even with help, even with financial aid, my aunt and uncle, it’s been a lot of money for them.
Not every family experiences difficulty related to access to health care, but often those who are themselves fortunate in this regard express concern for others who do not share their situation. For example, one father told us he had no problems himself, but knows it is a big problem for other people to get coverage. Furthermore, he isn’t worried about his child’s future because he expects a cure for CF within 10 years. This hope for a cure, which some families expect much sooner than this father, is one factor that counterbalances concerns about health care coverage. The belief that we are on the verge of curing the disease was most widespread in the year or two following the identification of the gene for CF. Much of that optimism that was always limited to cystic fibrosis families, and extremely rare among sickle families, has since turned to disappointment and even cynicism about medical predictions.
Chronic illnesses such as cystic fibrosis and sickle cell tend to keep adults from achieving economic independence. In the case of sickle cell, the economic consequences seem to be devastating for the families of origin as well. Fewer of them have the resources to achieve or maintain a middle-class life style while coping with the disease in a child. Mothers of children with sickle cell are more often single, and if married, their spouse’s income is more likely to be small. Therefore, loss of maternal employment is a more devastating economically. It does create eligibility for Medicaid, however, as one mother explained,
She’s on SSI . . . She has full coverage and benefits. The only thing I worry about is if I get a job and I get all of that assistance, then, they probably won’t cover my baby because it’s a preexisting thing. So that’s the only thing. Right now, I’m not complaining because the state is helping me and they take care of her and she gets all the medical care she needs. . . . But one thing I don’t care for is that I think she’d get better care if I had more money or better coverage. I think she would get better care and more individual care. Although I love Community Hospital and I love the clinic. . . I would like to have my own private doctor, a hematologist doctor, plus be affiliated with Children’s Hospital. But I would like to get her own doctor so that we can get a relationship established. . . . I don’t want all these physicians looking at her. I just want one person.(75)
The overwhelming majority of adults with SC are on Medicaid and disability and receive medical care free of charge. In the large urban hospitals from which most of our respondents were recruited, sickle cell patients who are seen at a sickle cell clinic may actually get better care than other family members without sickle cell, because they become known to the staff that specializes in their disease. In such cases, their access concerns have less to do with payment than with mistrust of medicine in general. While they tend to find individual providers they trust, there is a pervasive mistrust of the system as a whole. In a focus group with five lower class men with sickle-cell disease, there was discussion of the possibility that a cure exists for sickle cell disease that is being withheld, as well as concern about being used as “guinea pigs.”
I don’t trust the government to want to do anything for black people. Even like you [another man with SC in the group] said earlier, they may have a cure for it now, who knows. But it doesn’t mean that they’re gonna give it to us.
A third man agreed, saying, “I know . . . how secret and covert the government can be. I believe they’ve got a cure for it.” All of these men had found providers they trusted, but both black and white providers established this trust on a personal level. These personal relationships made it possible for sickle cell patients them to function within the health care system, but did not change their view of the medical system or “the government.”
In general, as we move up the socio-economic ladder within the African-American community we see more hope and less fear, but it is our view that very few African Americans believe race is irrelevant in determining the resources devoted to sickle cell research and treatment. With few exceptions, we received clearer statements of such critical positions when research team members who were themselves African American conducted individual interviews and focus groups.
It is difficult to determine the precise extent to which fears about loss of access to health care coverage are a barrier to genetic testing. Nevertheless, we have been able to identify several dimensions of the issue. Fear of being identified with a genetic disease, largely because of the chronicity it implies, creates a general atmosphere that is inhospitable to genetic testing for carriers. We find that it prevents diagnosis of disease in cases that are not severe, as well as inhibiting carrier testing. These are issues that deserve more attention by researchers.
Social and Cultural Differences Between SC and CF Families
Three issues emerged from our interviews as central to understanding the African-American response to sickle cell testing. To a great extent they also distinguish these families from the mostly white families confronting cystic fibrosis. The themes are: (1) Risk as Stigmatizing; (2) Zones of Relevance, and (3) Mistrust of the Neutrality of Medicine.
[footnote: While each of these themes can also be found among some of the white families at risk for CF, in this context, they are neither as widely shared nor as strongly felt.]
Risk as Stigma – The Redefined Social Bond. When medical professionals encourage or offer genetic testing or other services they usually do so using the language of risk. As with all terms, the meaning of the term “risk,” is a function of context and the context. Biomedical professionals have a sharply different understanding and use of the term than do the laity. This is particularly true among African Americans at the lower end of the socioeconomic ladder. Unlike a sense of danger that often arises directly from human experience, risk, as used in insurance and medicine, is an idea deduced on the basis of probabilistic and sometimes abstract calculations (Castel, 1991). It is most salient for those who have managed to minimize or eliminate the sense of constant danger and unpredictability from their lives. Historically, the concept of risk gained cultural currency among the middle class because “lowering risk” was used to create a sense of security, solidarity and community, linked as it was to the emergence of insurance and compensation for loss. Today, however, from the point of view of many African Americans the language of risk is associated with blame, stigma and exclusion. The term is much less threatening to white middle- and upper-middle class families who are more accustomed to confronting “risk” and then coming to believe that they have successfully reduced the risk in their lives.
We can see the differences in perception in the accounts of our respondents who describe not statistical risks, but instead speak of individuals as being “risks”. For example, “They told me I was a risk . . . ” This usage, equating individuals rather than probabilities as risks reflects recognition that the social bond has been redefined. Exclusion based on genetic vulnerabilities becomes a new social practice. Instead of defining the social group and creating mutual support, risk is now used to create and isolate the “at-risk individual”. Kenen (1995) has placed the emergence of the concept of the at-risk health status in historical context, pointing out that once acute illnesses were under control, the medical community expressed more interest in chronic illnesses and extended its control over formerly non-medicalized conditions such as pregnancy and childbirth. She suggests that the new at-risk status appeals particularly to the upper-middle class (p. 7) and the corporations and medical institutions that cater to them, as preferred clientele.
The use of the concept of risk to label and confer blame on the less privileged segments of society is a result of activities of specific interest groups who benefit by this interpretation. In contrast, when we look at how members of families with genetic disorders such as sickle cell (and sometimes even cystic fibrosis), use the term risk, we find that typically it is not a quantitative or mathematical term but simply another word for danger. When risk is used to refer to an undesirable possibility, the “risk” of genetic disease is no more likely to be a central concern than are many other potential problems such as loss of employment, loss of health insurance, or loss of a relationship. In our attempts to elicit narratives, the word often did not come up at all. Sometimes risk entered the dialogue when the interviewer used the word as part of a probe or follow-up question. When the interviewee introduced the concept, he or she was often quoting a provider, such as “I was told that if we both were carriers there would be a 25 percent risk that our baby would have sickle cell disease.” However, even well-educated interviewees often indicated their own precise risk figures were not especially relevant to them. As Abby Lippman’s (1979) work indicates, the major issue was simply the possibility that a child would either have an abnormality or it will not.
At the lowest end of the social hierarchy another meaning of the term risk is notable; risk as a “stigmatized identity.” For example, one woman told us, “The insurance companies don’t want to cover you because you are a risk.” or as another woman explained, “I can’t even get burial insurance . . . no kind of insurance . . . because you are too much of a high risk.” In this quote as in a number of others that refer to people as risks, interviewees use the term in the third person even if they have to switch mid-sentence.
A woman in her 30s with sickle cell disease told us that her fiancee’s family describes her as “a risk.” She talks about the difficulty she had telling people she had sickle cell disease and how this resulted in them thinking she is a risk. In her experience people call her that when they want to prevent her from becoming a wife or mother. Her response to this situation has been to have four children, two of whom have sickle cell disease. She takes pride in her ability to prevent and respond to her children’s crises without having to resort to hospitalization. In this context her experience is valuable and she is no longer stigmatized. She found a way to triumph over the perception of herself as a risk, which is to have children with sickle cell who can appreciate her expertise in dealing with situation.
Overwhelmingly our respondents reject prenatal diagnosis and selective abortion as a strategy for determining which pregnancies will be brought to term. They accept abortion but reject selective abortion.
This definition of risk may be related to the fact that as Lisa Handwerker (1994) has shown, since 1987 the courts have increasingly used medical testimony to prosecute women labeled “high risk” for failure to comply with medical advice when their fetuses or babies die. She suggests that attempts to prosecute women discourage rather than encourage seeking medial care. In spite of the fact that the assessment and management of risk for many conditions is neither standardized nor consistently applied, health care providers and the legal system, unfortunately, make decisions as if risk were objective “fact.” By contrast, patients understand risk as part of their complex and ambiguous social world. They are well aware that the label itself may confer misfortune, and the benefit it could confer is much more obscure and remote.
Zones of Relevance. Closely related to the concept of risk is the issue of relevance. The more predictable the environment, the more attractive it seems to be to engage in the formal risk analysis that genetic medicine often requires. Phenomenologist Alfred Schutz noted the fact that different things carry a different weight for people at various times; he called this notion “zones of relevance.” The most attention is given to what he calls the “primary zone.” In certain situations, it is in these areas that we most often solicit expertise (Schutz, 19??:124). Asking for the help of experts requires confidence that the specific danger being focused on has been selected for one’s personal as well as the collective benefit. This confidence decreases, however, as one descends the socio-economic scale.
In talking with one father in our study, we see that he rarely has the occasion to put sickle cell first. Besides his own children, he has taken on two of his brother’s sons, ages 10 and 13, whom he describes as “needing some structure.” He has been unemployed for the last year and is constantly trying to create work for himself while his own sons are both entering “manhood” and facing confusion about their own futures. His oldest son’s severe case of sickle cell disease is only one of many problems, but the one for which most assistance is available. He explains:
There’s an old proverb that [says] when your house is on fire you don’t think about broken windows. In a lot of communities where sickle cell is [present], they’re struggling. People are very poor and they have a lot of problems so they don’t look at this particular problem as being one that’s overwhelming in relation to other problems that they have.
Thus for families such as this, genetic information has little relevance until it comes to the fore during a specific episode of sickle cell crisis.
When an uneducated young woman fails to remember her precise risk figures, or doesn’t appear to be taking them as seriously as some might think she should, she may be engaging in a much more complex calculus of multiple risks. One of those risks is the possibility of not finding love or bearing children at all. As one woman told us, “lots of kids have problems . . . I wouldn’t let that [the risk of sickle cell] be an issue.”
The selectiveness of medical constructions of risk does not escape those who view life as an endless series of challenges. Furthermore, it threatens one area of life which, even if only temporarily, offers relief from a sense of failure, romantic love. The importance of such relationships as a primary zone of relevance and thus a source of resistance to carrier testing must not be overlooked. Respondents frequently indicated that they perceived the calculatedness of risk analysis to be incompatible with emotional commitment, as illustrated in an earlier cited quote linking genetic testing with dog breeding. We found very few African-American women willing to adopt a scientific or instrumental rational approach to their own pregnancies. This is partly because pregnancy represents a commitment to a relationship that is recognized to have multiple perils and more immediate perils.
Mistrust of the Neutrality of Medicine. The markedly precarious economic situations of most African-Americans is what makes both “risk as stigma,” and “zones of relevance” salient in their responses to genetic testing. Another factor that is perhaps more deeply rooted and that is less clearly class-related is mistrust of medicine. African-American respondents gave many indications that they mistrust the neutrality of medicine as an institution and thus are often less likely than our non-African-American respondents to view it as a source of support. Their narratives are peppered with references to medicine as more a tool of domination and control than one of health care providing. This is particularly striking in comparison with the narratives of the predominantly white cystic fibrosis families. An African-American family is much more likely to have had their child’s symptoms considered to be the effects of child abuse or to have relatives and friends who have experienced what they consider to be medical abuse. The widespread discourse of mistrust is often affirmed through repeated encounters of direct personal experience. A woman with sickle cell reports that an army doctor who diagnosed her as a child warned her mother
“They like to do a lot of tests on sickle cell patients.” And he told my mother, “don’t let them do that to your kids because they don’t have a cure. They’ll promise you that they have one and they don’t have one.” And so she was worried about letting them do anything to us . . . So she just old us, if you’re getting medical attention, make sure . . . you’re not just being a run-of-the-mill guinea pig. (363)
This concern about being used as “guinea pigs” arises with particular frequency, among African Americans. The field of genetic medicine is a particularly sensitive area in this regard. One father of a child with sickle cell disease told us:
I don’t have a whole lot of trust around the way the whole medical field operates, and there may be some very well-intended people out there . . . but, you know, the problem is now that things are becoming very polarized around issues that are coming up. . . if you don’t know your history, you are doomed to repeat it. . . . they had folks during WWII and before who were trying to create a super race and people who view genetic weakness—and what does this mean for the country? . . . I think all that comes into play. . . We get used as cannon fodder sometimes you know, we’re throw-aways. I think there’s always been doctors who tried to prove that Black folks were inferior.
Among those closer to the bottom of the economic ladder, particularly among Black men, we find a very explicit belief that medical advances are unlikely to benefit interviewees or anyone in their community. A man with sickle cell disease told us:
I sincerely do believe that a lot of the diseases that are out right now . . . the government does have cures for them. I know the power does. . . I believe they’ve got a cure for it . . . If that happens, the FDA is gonna lose a lot of money, the pharmaceutical companies is gonna lose a lot of money, doctors . . . Because they wouldn’t need Motrin, Ibuprofen, and a lot of other transfusions and things. They wouldn’t need a lot of that stuff. . . . People with sickle cell anemia, they do pop a certain amount of money into, I mean, I’ve spent my share to get those forms of help. If I have sickle cell anemia and I have a cure, I don’t need those things no more. That’s gonna be a couple thousand dollars lost by me alone. Imagine the other people. I don’t know what the percentage rate is, as far as finances. It’s up there. It’s in the millions.
This distrust is sometimes associated with personal values of providers, but generally African Americans understand racism in medicine as a result of larger social and political processes. Often they see problems with science as extending beyond race to values associated with power in general. Even when medicine is seen as having benefits, suspicions still remain — and more consistently so among African Americans than among the white cystic fibrosis families with parallel low levels of education.
Modern medical technology and all this good stuff has its good points. A bone marrow transplant can help a person—great! [But] if Tyrone needed a bone-marrow transplant, I would really have to talk to a half dozen people to give me some viewpoints. . . I want to know those risks, and I want to know the side effects, and I want to know the chances: “How many times have you done this and what are those results?” I’m going to grill somebody. I won’t be sitting up there blind, being clueless as to what’s going on.
This mistrust has its roots in events African Americans often cited such as unauthorized sterilizations of black women, the use of genetic testing as a surrogate for racial discrimination, and the experimental research rationale used to withhold treatment of Black men suffering from syphilis in the Tuskegee Syphilis Study. The latter is an atrocity for which the U.S. government has recently formally apologized (Los Angeles Times, 1967). More recently this mistrust has been fueled by attempts to identify poor blacks with a biochemical inclination for violence and by the contested claims of the link between race and the measurement of “intelligence” by the authors of The Bell Curve (Hernstein and Murray, 1994). This is the context in which responses to the concept of genetic risk can be better understood.
While both men and women give evidence of mistrust of the institution of medicine, African-American women are more likely to focus on individual providers and are thus more willing to attempt to actively negotiate the world of biomedicine. They are also more frequently able to forge personal relationships of trust in clinical settings.
Societal Implications
The final difference we will discuss between sickle cell and cystic fibrosis respondents is the difference in their perceptions of the social implications of genetic testing. African Americans, in particular, are concerned about potential abuses of testing in the social and political realms as well as in medical practice. This theme is much more pervasive in interviews of both African-American men and women than among whites. A good example of the general mood on this topic was expressed by a mother of a child with sickle cell:
You know, if I were confident that genetic testing would be used for the proper and righteous reasons, I probably would not have a problem with it. But I know that is not the case, and therefore, I do have a problem with it. I know people will be discriminated against. I know that! We can say anything we want, but the facts are, have been and will be that there will be discrimination. There will be unfair treatment of a lot of people as this becomes the rule of thumb, or the order of the day when it shouldn’t be like that. I can see people even being divided up into groups. You know, you got all this, you can’t do this—you know, dictating to people what they can and cannot do. And I think that is just wrong. I don’t like it. I’m not comfortable with it because I don’t think our society is mentally capable of handling it properly.
Her comments are representative of many African Americans and are less cynical than many. Yet this concern is not limited to African Americans. A white woman whose sister died of CF, but who describes herself as strongly pro-choice, expressed feeling threatened by the social values she perceives that are implicit prenatal diagnosis and selective abortion.
I really rebel at the thought of fetuses being aborted because they are not perfect. There is just something heinous and barbarous about that in my mind. And I don’t feel a prejudice against a woman who might make that decision, but I do feel a certain level of contempt for a community that would try and support that kind of thinking, because it is so fascistic that it makes me sick. . . I don’t feel that kind of effort can be supported without supporting the idea that kids with disease are mistakes, so I would only have an inclination to think it is acceptable if there is equal support of people’s right to be different, to be disabled and to be ill.
While the level of concern and mistrust is definitely lower among whites, a distinct lack of enthusiasm for genetic testing is apparent in most interviews. The dimensions of molecular genetics that both CF and SC family members find compatible with their own social worlds, their priorities, commitments and perceptions of reality, are treatment and cure. They appreciate advances in treatment and hope for a cure with varying degrees of optimism. When we asked members of CF families if they followed developments or advances in genetics we consistently got hopeful responses about new treatments, particularly the possibility of gene therapy. Sickle cell family members are less optimistic, which is not surprising given that carrier testing has been available for decades but few new treatments have emerged. Rarely do interviewees express enthusiasm for screening or testing— even when they make use of them.
To the extent our interviewees are optimistic about a cure on the horizon, they have less enthusiasm for genetic testing. Ironically, faith in science’s potential to cure the disease may be one of the major barriers to testing. For example, one aunt of a child with CF told us:
With the advances that they are doing now, it could be a whole different decision—one that it falls in line as a minor problem.
Whole families are alert for advances in treatment. Without exception, when asked if they follow advances in genetics, family members comment on advances in treatment, not in testing. A grandfather interviewed in July of 1993 told us:
I fully believe that they will have within, I would say, by the middle of next year, a year from now . . . I think they are going to have a cure for cystic fibrosis. . . I fully believe that within a year they will be on top of it. Just like infantile paralysis. Our research is fantastic. It is just great.
While each disease group has distinctive patterns of dealing with the disorders, we have been struck by similarities in resistance between families in which cystic fibrosis has been diagnosed, which are overwhelmingly white and those in which sickle cell disease has been diagnosed, which are overwhelmingly black. Both groups of families make an important distinction between medical knowledge directed toward care, and genetic testing for the purposes of “prevention.” Both welcome the care-giving aspects of medical knowledge, and both display resistance and avoidance to testing for prevention. Thus a common response to prenatal diagnosis is to use it, not in relation to any decision about continuing or terminating the pregnancy, but for reassurance or to provide an early warning system should the fetus be affected. One woman explained that it is better to get the grieving over the loss of a healthy child over with before the baby is born. Using prenatal diagnosis to prepare the family, both emotionally and practically, is a more common strategy among CF families than sickle cell families, partly due to better access to early prenatal care and to health care services in general.
Thalassemia
A focus on thalassemia was not part of the original project design. It evolved primarily because one of our clinical settings offered a rare opportunity for access to patients or families affected by thalassemia, and because physicians at this hospital asked for our assistance in understanding the perspectives of these patients, many of whom are recent Asian immigrants to the U.S. We were eager to study these families, because like sickle cell disease and cystic fibrosis, thalassemia is a life threatening recessive genetic disorder and this group could provide us with yet another level of cultural and social variation on how high-risk families relate to genetic disorders and genetic testing.
[footnote: Thalassemia major (the most serious form) negatively affects the production of red blood cells (hemoglobin) such that the tissues have insufficient oxygen, thereby leading to anemia. Monthly blood transfusions are standard treatment, and the need is usually apparent before 18 months of age. At this age, the child often has signs of anemia, stunted growth, or an enlarged spleen, also manifested in a swollen stomach. Without treatment, the bone marrow (the tissue inside the bones that forms red blood cells) expands as it tries to manufacture more of the necessary red cells. This development weakens the bones and causes bulging in the cheekbones and forehead areas. In addition to these visible changes — facial distortions, prominent bone structure, short stature — there generally is also loss of appetite and increased irritability. Bone marrow transplants are the only long-term cure for this disorder, and are much more likely to be successful with children under seven than older age groups. They are least risky with a fully compatible sibling match, although new technologies are making unrelated donors a possibility for the distant future. A person with beta-thalassemia major must, at puberty, begin desferal treatment, which works to remove excess iron which accumulates in the body as a result of regular blood transfusions. Because of advances in treatment, the issue of life expectancy is largely indeterminate, although life expectancy is clearly increasing. ]
Methods. The respondents from families affected by thalassemia, unlike the CF and SC respondents, were all recruited in a hospital setting. We had hoped to move beyond this initial point of contact into the extended families but this was rarely possible given time and resource constraints. Thalassemia interviews faced the same difficulties we encountered with SC and CF families, such as time pressures on parents and the need to care for children. However, we had to overcome additional obstacles as well. These included language barriers, a cultural reluctance to discuss personal matters with outsiders who were not medical professionals, and feelings of vulnerability due often to tenuous residency (immigration) status.
We examined a total of 25 cases (parents and affected adult children) representing a variety of middle-eastern and Asian cultures. The sample of Asians affected by thalassemia was the largest subgroup, and the group in which we began to identify culturally patterned responses. Yet, the Asian sample is diverse with respect to ethnic background (e.g. Chinese, Vietnamese, Laotian, Cambodian) and religious orientation (Catholic, Buddhist, no religion).
Overview. Because only members of nuclear families were interviewed, the range of familial responses has hardly been tapped. Nevertheless, we have identified some interesting issues, and we will briefly summarize those most relevant to genetic testing.
Responses of family members can be broadly categorized into two types: (1) avoidance, expressed as reserve, distance, and minimal communication, and (2) vigilance, expressed as interest, energy, and engagement. These patterns of vigilance and avoidance need not be mutually exclusive, and it is apparent that some of these experiences may reflect “stages”, either linear or cyclical. Vigilance occurs when people attempt to reduce uncertainty by seeking knowledge and acting on that knowledge. Avoidance is a strategy to attempt to cope with uncertainty by blocking, ignoring, or circumscribing unpleasant knowledge (Weitz, 1994: 138-139). Several factors associated with thalassemia seem to foster “avoidance” patterns, while other factors encourage vigilance among these families.
[footnote: Any one interviewee, however, might manifest both patterns, being distant and avoiding in one context (e.g. hospital) or stage of illness (e.g. early diagnosis) while vigilant in another (e.g. seeking out information about alternative cures).]
Patterns of Avoidance. Parents must first come to terms with the diagnosis. A typical initial reaction to the diagnosis is shock and resistance. Thus, one research assistant recorded in her notes the strategy one of the medical doctors at tje hospital routinely tried to break through parental resistance to starting blood transfusions:
What he does is let the kids get pretty sick so the parents can see with their own eyes that the kids need help. Then he starts the transfusions and lets the kids deteriorate again. This proves to the parents that the kids benefit from the transfusions — need them to be healthy. Dr. Jones feels this will help families coming in to be transfused. Dr. Jones said that he knows from a kid’s blood levels far in advance when the kid is going to need transfusions. But he can’t just start giving the kids blood right away before the child gets sick because then the family wouldn’t believe the kid needed transfusions. “I have to do it that way because families have to buy into the treatment.” Dr. Jones seemed to feel that families had to see their kid was very ill before they would commit to anything so arduous as monthly transfusions.
In the early stages of diagnosis the parents search for meaning. The question, “Why my child(ren)?” dominates, as individuals try to absorb this personal sense of crisis. Genetic knowledge alone is of limited value in promoting “acceptance” of the disorder at this level, even if parents accept the fact that the disease had to do technically with “something in the blood.”
A major cultural belief competing with the dominant or official medical understandings of disease is a belief in karma. A Chinese father of a thalassemic child (351) was described by the interviewer as still having to cope with his own contributing role in the disease, a culpability associated with another lifetime : “He used to blame himself for the fact that (the child) has Thal major. He sometimes thinks that he did something bad in another life and that now he is paying for it by having a child with problems.” Similarly, a Chinese mother (364) at first didn’t tell anyone in either her family or her work about (the child’s) thalassemia. This seemed to be a combination of not wanting to make her relatives sad and worried about (the child) and also realizing that they might extrapolate from this that there was something bad in their family that they had done to deserve this. One pattern discerned was a cultural belief that imperfect children are punishment for bad associations in the past—even going back several generations through the ancestors.
There is also a comtemporary version of culpability. As with sickle-cell anemia and cystic fibrosis families, the medicalization of illness does not always effectively neutralize feelings of blame. Mothers especially may experience guilt and feel that the disease is a result of something they did wrong during pregnancy (316, 331, 359, 408). About his wife’s long-standing inability to accept the diagnosis of their thalassemic child, a Chinese male (359) reported:
At least she isn’t crying anymore, and she for the longest time for about 3-4 years she cry, you know, when she sit and alone, and I can, I caught her crying. And then we talked about it… she’s now little bit more fatalistic in some ways. I think it’s, it’s difficult for her to accept… does understanding of a phenomenon helps one to accept its, its, condition? I don’t know. I’m not trained in that field, so I really don’t know. Ah, I think she has enough understanding of the thalassemia as a disease or disorder, but does that mean any easier for her to accept the disorder when it happen to your own child? I don’t know.
Some of the self-blame in the above instance is attributed to guilt for not recognizing that a series of miscarriages prior to the birth of the child could be signs or warnings that something was amiss.
Another mother of a thalassemic child (333), an immigrant from Thailand, worried that when her eight-year old son gets older, he will ask questions about his disease and blame his parents. She is not otherwise ashamed to talk about the disease and, in fact, is glad to inform others about it. Furthermore, precisely because thalassemia and genetic ideas are not part of general public knowledge, other family members may also resist testing for themselves. The avoidance of diagnosis or testing has implications for screening. Commenting on this, a researcher recorded in her field notes:
Mrs. X’s in-laws, however, did not believe this explanation. They thought that it was all Mrs. X’s fault that she and her husband had a sick child. Her mother-in-in law in particular blamed her alone for the problem saying that her child was sick because she was so skinny. Mrs. X also said that her husband’s brothers and sisters did not believe that it was a disease that ran in their family until three years later when her husband’s brother had a child with Thal. At this point her husband’s younger sister went to get tested before she got married. (316)
Another case is of particular interest because it shows how major shifts in cultural attitudes can nevertheless occur with both medicalization and shifts in social contexts. This pattern, which was surprisingly common among those we spoke to points to reorientation from avoidance to vigilance. The shift is most clearly delineated, however, in this example. The father of two thalassemic boys (331) reported that in Hong Kong the stigma associated with a genetic trait would ruin one’s chances of marriage, constituting a major barrier to genetic testing, even though the Chinese Family Planning Association was said to encourage all citizens to take such preventative measures. Attitudes towards treatment, in turn, reflected a form of cultural fatalism. Thus, regarding this father’s own situation, the interviewer wrote:
Initially Mr. Ho was reluctant to have his son transfused because he held the belief of traditional Chinese philosophy that things were the way they were meant to be. He felt that treatment would only extend his sorrow. His son, however, began to receive transfusions and after that Mr. Ho changed his mind about the treatment doing more harm than good because it was obvious that the child felt much better when he was being transfused.
The shift in attitude, noted above, eventually extended to not only accepting the genetic definition but to advocating testing for all kinds of problems, such as Down syndrome or other developmental disabilities.
While fatalism can be rooted in cultural beliefs, whether this attitude is reinforced or overcome depends similarly on the social context of the experience of illness. Thus, the mother of a 15-year old boy who was diagnosed in Hong Kong as thalassemic (316) reported that her son was now much more optimistic about his condition since arriving in the United States, primarily because he saw for the first time that there were thalassemics older than 30. He concluded that he would not necessarily die young, but conceivably grow up and even marry. By contrast, a 15-year-old Cambodian patient was described as pessimistic about her future, largely because she saw her older thalassemic sister not doing well. Her missed hospital appointments and noncompliance with treatment schedules might be seen not only as “self-fulfilling prophesies” but as an avoidance reaction to her own image of the prospects which awaited the “inevitable” unfolding of her own condition. (404)
In some cases, vigilance was undermined by or diverted by social problems not directly related to the disease itself. Thus, a Cambodian mother was distressed over her son’s gang affiliations, delinquency, school problems, and running away from home. As the researcher or field worker noted, she was “not sure whether his problems are because of his Thal or because of his being a teenager.” (405)
Patterns of vigilance. While Asian health beliefs contain certain elements of “fatalism” in their orientation, it is also true that there is a great deal of activity based on notions of effort or “willpower” towards overcoming problems. These attitudes seem relevant to explaining the greater propensity towards vigilance as they encompass specific medical, historical, and social/cultural factors which not only converge to produce this more vigilant frame of mind, but which also differentiate the thalassemia population from the other two risk populations.
As recent immigrants, this risk population encounters the American health care system for the “first time,” and therefore, their experiences are, by definition, less predefined. Whatever cultural beliefs may have competed with the initial medical diagnosis, thalassemia families on average are less mistrustful than sickle-cell families towards what Western science has to offer. The possibility of receiving a bone marrow transplant is perhaps the best indicator of this receptivity. The “non-compliance” and missed appointments reported by physicians, on the other hand, seem to stem largely from relations to an impersonal hospital bureaucracy, the strict requirements of the treatment regimen that is to be sustained for life (i.e. the administering of desferal injections five times a week), and the fact that patients generally do not feel sick enough at the time they are expected to return for treatment (402, 403, 405).
Alternative Medicine. Another reason that might explain the propensity for many thalassemia families to be “vigilant” is the relative availability of a wider range of health solutions or treatment options. While sickle-cell and cystic fibrosis families also explored nonconventional avenues, it is even clearer in the thalassemia cases that families draw upon their traditional cultures. As with the sickle-cell and cystic fibrosis families, a college education heightens the tendency to assume greater responsibility for health care. This tendency will take an “alternative” turn in the case of thalassemia families, where there are strong cultural ties to a distinct medical tradition. In general, class differences do not appear to be obstacles to pursuing these alternative health care strategies, because these alternative approaches are often less expensive. Since our recruiting of respondents occurred in a hospital setting, it is not clear, however, how much they rely on such alternative avenues of care to the exclusion of Western medicine, or use it conjointly with hospital care.
Grandparents in one household (364), for example, played an important role in seeking alternative medical treatment, taking their then two-month grandson to China to confirm the genetic diagnosis. The maternal grandmother visited a number of hospitals there, consulting different doctors in order to better understand the problem. Altogether, the child stayed in China for six months and received treatment for thalassemia via a somewhat different method. This involved a combination of more frequent transfusions, acupuncture, and iron pills. Despite the fact that the maternal grandmother belonged to a household of fourteen, where the total family income was under $30,000 annually, economic resources were reported not to have been a significant barrier. As the mother explained, the cost of having her child tested and treated in China amounted to only $300 in Chinese money or $50 in US dollars. Eventually, it was the low blood supply and the lack of individual medical attention (1 nurse for every 20 children) that influenced the family’s decision to bring the child back to the United States for treatment.
While economic resources (including insurance coverage) enable the consideration of other medical options (such as bone marrow transplants), educational level seems most critical in shaping how individuals respond to or integrate medical information into their lives. It may be that the high value placed on education in Asian-American families with thalassemia contributes to this willingness, interest, and ability to independently pursue medical knowledge.
One Chinese father (359), for example, pointed out that he came from a family in which everyone was college-educated. As an engineering manager who earned over $50,000 a year, he took an extremely rational and intellectual approach, avidly reading a range of medical and technical articles, in Chinese-language sources as well as English. While traditional healing practices were investigated along with advances in Western medicine, the former were eventually terminated because progress could not be objectively measured. As he explained, the process was “to a large degree like rolling the dice, but you are, you are in the blind. You don’t know whether there’s any progress made, or if there’s any slip back being made, because there’s absolutely no way of monitoring it.”
Bone marrow transplants. As in sickle-cell and cystic fibrosis families, chronicity presents an inevitable drain upon financial resources, concerns about insurance coverage, the effect on a potential mate, and certain problems with long-term care. In the case of thalassemia, blood transfusions at the hospital and desferal treatment at home lead many to pursue the possibility of bone marrow transplants, which promise far and away the greatest hope for long-term cure. Without this procedure, medical complications can result with the development of antigens, which in turn would lead to the rejection of blood transfusions. The same father who had explored traditional healing methods only to rule them out as inadequate because progress could not be objectively measured also felt that transfusions were not the long-term solution.
…my wife and I still hold the view that transfusion is not a long-term solution. Although it alleviates the symptoms of thalassemia, blood transfusion is not a, solution, not a true solution. The basic disorder still persists. As she grow older, given the frequency of transfusion she will run into some point in time, . . . having too many antigens in her body. That she will start rejecting transfusions, so to us that is not a long-term solution. I also understand that at that point in time, the bone marrow transplant was the true solution. If it works then she would haven’t to do that again…(359)
While bone marrow transplants are expensive procedures, his own cost-benefit analysis (comparing the cost of hospitalization for this procedure with the cost of regular transfusion and other related medicine) indicated that the family would break even in five years.
While there is a waiting list for possible bone marrow donors, the ideal donor is likely to be a sibling, though in one highly unusual instance it was the mother (355, 356). Because of the heavy burdens of raising a thalassemic child, some parents do not want to have any further children, especially if there is this risk of the child having thalassemia and abortion may or may not be a viable option (333, 351, 405). Others are undeterred by the concern of having additional children and see this as a real strategy by which to find a possible match (364, 410). Thus, a Chinese mother of three, who lives in a household of 14 other family members (364), indicated she had gambled on having a second son in the hopes of a match. While this son was a carrier, not a match, her third child, a daughter proved to be match.
[footnote: When both parents are carriers, the mathematical probabilities of inheriting Beta-Thalassemia major are one in four, which in genetic terms, refers to the chances of both parents passing on to the child an abnormal (altered) genes for hemoglobin.]
In short, although genetic science can aid in monitoring or ensuring optimal care post-natally, Asian families with thalassemia individuals are predisposed towards taking a more proactive stance. This is reflected in efforts to take advantage of prenatal testing, as well as search for a bone marrow match.
Abortion. Since thalassemia is like other recessive diseases, its diagnosis is most frequently post-partum and the first time that parents have ever heard of the disease. For this reason, abortion was rarely an option in the families observed and interviewed. However, when abortion was suggested as a hypothetical possibility, there was little hesitation, reflecting a practical stance. While there were also exceptions (333), including a woman who worked at an infertility clinic (368), family members generally did not hesitate to say they would have chosen abortion had prenatal information been available. Having a chronically affected child merely reinforced these attitudes. This is in marked contrast to cystic-fibrosis and sickle-cell families. In general, abortion was considered by Asian immigrant families to be a viable option. Their views on this topic seem to be relatively unaffected by religious notions, but perhaps more importantly we hypothesize, it may reflect cultural differences in the definition of the fetus. There is little indication in our interviews of the level of imputing personhood or equating human life with the fetus, as we certainly found in families at risk for CF and sickel cell anemia.
In one rare instance the husband and wife apparently had the opportunity to abort because the disease was diagnosed beforehand. They chose to not abort. The respondent who was the father (355), indicated that family members were surprised that the pregnancy was not terminated: “Several people have said why you keep — have her? Why you have her?” Ignorance about how serious and debilitating the disease may have been one factor, since he did reflect retrospectively that abortion would have made better sense given the incredible burden of living with the disease.
AIDS. Although shame and social isolation have been among the culturally traditional responses to certain illnesses thought to be contagious, hereditary, or stigmatizing (e.g. mental illness), the major social avoidance patterns connected with thalassemia have to do little with the genetic disease per se, but rather are related to risks that blood transfusions seems to pose in terms of contracting AIDs. Given that treatment typically consists of monthly transfusions, other family members were concerned about social contact and exposure to the AIDS virus. The following case illustrates how social isolation was more emotionally wrenching than the genetic disease itself, complicating relationships around the primary medical condition.
Recalling how his own immediate family was placed in pariah status by relatives fearful of contact and contagion and therefore “openly hostile,” one father (359) described this period as “the most difficult phase in my life.” Although family members eventually “started accepting her (the thalassemic child) back” when AIDS information became more accurate and widespread, the social rejection was particularly hard on the wife because it was a Chinese family, where ties that generally exist between sisters (now distanced by the fear of AIDS) are usually closer than those between brothers. The husband thus recalled this as a period where his wife “cried a lot,” and the two took “extremely long walks” to ease the pain of social isolation.
Because genetic diseases, unlike AIDS, are not perceived as “contagious,” the medical diagnosis can relieve a great deal of guilt or blame. The same respondent thus explained that apart from that critical period clouded by the fear of AIDs, parents, friends of his parents, and acquaintances are now quite accepting of the daughter and her condition. The contagious versus non-contagious aspect of illness is central:
I don’t think there’s reason for them to behave any more differently than they normally do, do now. Will they reject us because of (the child)? Ah, I don’t know. I mean, we — it’s just like, you know, if one of my friends have — their parents — are arthritic, do I reject them because of that? No. It’s not contagious disease. I don’t care. I mean, if it’s contagious disease, that may be different. It’s not contagious disease, so I don’t know. I don’t think so.
In general, to the extent that patients did not “talk about” the disease to others, there were at least two main reasons: (1) the fear of social rejection as a result of HIV status, the threat of HIV, or the consequences which either HIV or genetic status might have on employment (351, 359, 364, 392, 401, 402, 403), and (2).
Once the definition of the problem as a genetic disorder is accepted and understood, thalassemia often evokes a proactive attitude towards genetic testing and communicating such information to family members, as well as a more favorable attitude towards abortion than we observed in either the sickle cell or cystic fibrosis respondents. Language barriers currently are a major barrier to many being able to fully absorb genetic information or the medical advice that is related.
Based on these preliminary findings, it is likely that public education about thalassemia and genetic testing would be well received, particularly given the preventative orientation of this population and its willingness to incorporate genetic knowledge into their reproductive planning and behavior. Genetic knowledge can function to promote more “rational” communication in thalassemic cases, but the medical definition does not entirely eliminate family tension or stigma.
It has been noted elsewhere that unquestioned acceptance of medical authority can make issues of informed consent problematic: “The medical model is complementary to Asian expectations and respect for authority and may lead to uninformed consent to genetic professional’s recommendations” (Wang and Marsh, 1992: 81). To the extent that thalassemia is understood to have stages in the long-term unfolding and adjustment to its chronicity, the construction of meaning around the disease is likely to be more “open-ended” and responsive to new interpretations or understandings during those periods when some kind of medical adjustment is necessary (e.g. the initiation of desferal treatment). Further research, in other words, is needed to determine how cultural beliefs intersect with this definition at various stages of illness and disclosure, and the extent to which the adjustment process reflects a reconciliation of non-medical issues with the official diagnosis. The overall positive response to genetic screening can be likened, on the surface, however, to the positive response of Americans of Ashkenazi Jewish descent to Tay-Sachs screening, a useful point for any future comparative study.
Summary and Implications
This study has examined the processes by which genetic knowledge is integrated into the lived experience of high-risk family members. We have observed a wide range of responses to new genetic knowledge — from the kind of vigilance that proponents of genetic testing advocate to avoidance and even opposition. We have found that these responses to not occur randomly, but are patterned along social, cultural and economic dimensions. Among families at risk for cystic fibrosis compared with those at risk for sickle cell disease there are significant differences as well as interesting similarities. One major pattern we have observed transcends ethnicity or genetic condition. It is this: the closer people are to someone with genetic disease, the more problematic and usually unacceptable genetic testing is as a strategy for dealing with the issue. To understand the counterintuitive finding that enthusiasm for genetic testing is lowest among those closest to these conditions, we must remember that the successful functioning of high-risk families rests on their acknowledgement of the value of the lives and contributions of their members with genetic disease.
High-risk family members who support genetic research do so primarily because they view it as leading to better care and ultimately a cure, not because they support genetic testing.
Sustaining relationships in the face of the gene disorders we have described requires belief in a social bond that can withstand the threat of disability, as well as the recognition that the value of the life of a person is not diminished by physical limitations. These values are woven into the narratives that hold these families together. Without these values, relationships cannot be sustained. Genetic testing as a strategy for eliminating the burdens of genetic disease is experienced as a potential threat to these values.
When we began our study (in 1992), shortly after the late 1989 identification of the genetic mutation that causes cystic fibrosis, there was great joy and enthusiasm for genetic research among families with an affected member. A few respondents even cited the expectation of an imminent cure as justification for not being tested. Since that time we have watched this hope fade, but without any increasing enthusiasm for genetic testing. It is difficult for family members to integrate their personal and family narratives with those of molecular genetics when the latter is used primarily for genetic diagnostics rather than in a context of caring. As a result genetic testing tends to be most acceptable, and most easily considered, among those whose concepts of genetic disease are disembodied and abstract — those without a family history of the disease.
A major policy question related to genetic testing, the degree and extent to which consumers want expanded access to such procedures has been discussed without much empirical data. The assumption implicit in recent decisions of the Cystic Fibrosis Consensus forum (April, 1997) to offer testing to those with a family history, is one rooted in the social worlds of molecular and clinical genetics From this perspective, it makes most sense to offer the test where it is most likely to come up positive. Yet, this can only be effective if results influence reproductive plans. Our research indicates genetic testing rarely changes reproductive plans. If policies were to be based on the expressed needs of high-risk families a much greater emphasis would be placed on treatment and care. Testing would be de-emphasized.
If genetic testing is to continue to target those with family histories of genetic disorders, programs must recognize that the intra and interpersonal conflicts regarding genetic testing are much more complex and disturbing than heretofore considered. It is in these families that conflict, issues of guilt, shame and blame, and difficulties in integrating the new diagnostic technologies are most likely to exist. The practice of offering carrier tests or prenatal diagnosis to those with a family history of a genetic disease, is therefore, much more problematic and complex in terms of potential counseling needed. We therefore join a host of other commentators and ethicists in clearly stating that the practice of offering carrier testing or prenatal diagnosis to the general population is not a good idea (Holtzman, 1997, Task Force). We now have data from the lives of those “in the experience to support this position.
Racial-ethnic differences in rates of utilizing prenatal diagnosis, as well as responses of all potential consumers of genetic testing are best understood by exploring how the social and cultural contexts of those actions are constructed. These, too, are illuminated in the narratives of our respondents. Here we can see the frames of meaning lay persons draw upon in making sense of their lives. Central among all ethnic groups are norms and values that frequently run counter to the instrumental-rationality of the world of molecular and clinical genetics. As we have seen, on a continuum, African Americans are much more sensitive than whites to the potential misuse of science and medicine. As a collectivity, their caution is firmly rooted both in a culturally invoked history and in often stark current economic conditions. This caution is not likely to wane until these conditions are changed. Indeed, until African Americans can see medical institutions devoted to addressing a wide range of health care needs, individual practitioners promoting genetic testing will continue to be viewed with a far greater level of mistrust and suspicion than they can comprehend — given their own ideology of neutral technical advances in biomedicine and genetics.
Although more interest has been exhibited in understanding why certain ethnic groups appear more resistant to new genetic technologies than others, we believe the most important finding of our study is the similar difficulties all groups experience in integrating two divergent discourses. One of these, the discourse of molecular genetics, brackets the larger social context. Meanwhile, the nature of the social worlds in which high-risk families live is by definition context-dependent and negotiated using basically different domain assumptions. Where genetic testing is embraced as a useful tool in realizing one’s reproductive plans it is because family members have found a way to define testing as consistent with the personal and family narratives that form the organizing framework for their behavior. Often this means defining the well-being of existing children with disease in terms of benefits accruing from testing and even selective abortion. In any case, it is in a context of care that genetic testing is most likely to be successfully integrated into family life.
REFERENCES
Bellenger, D., K.L. Bernhardt, and J.L. Goldstucker (1976), Qualitative Research in Marketing. Chicago: American Marketing Association.
Bishop, Jerry E. and Michael Waldholz (1990), Genome: The Story of the Most Astonishing Scientific Adventure of Our Time—-The Attempt to Map All the Genes in the Human Body, New York, New York: Simon and Schuster.
Bowman, J.E. (1977), “Genetic Screening Programs and Public Policy,” Phylon, 117-142.
Bowman, J.E. and R.F. Murray Jr. (1990), Genetic Variation and Disorders in Peoples of African Origin, Baltimore, Maryland: Johns Hopkins University Press.
Castel, R. (1991), “From Dangerousness to Risk,” The Foucault Effect: Studies in Governmentality, 281-198.
Duster, Troy, D. Matza, and D. Wellman (1979), “Field Research and the Protection of Human Subjects,” American Sociologist, 14:3, August.
Fujimura (1997), “Canons and ‘˜Purity’ Control in Science: The Howl of the Boeotians,” conference paper, 119th Annual Meeting of the American Ethnological Society, Seattle, Washington, March 6-9.
Glaser, B.G. and A.L. Strauss (1970), “Discovery of Substantive Theory: A Basic Strategy Underlying Qualitative Research,” in W.J. Filstead, ed., Qualitative Methodology: Firsthand Involvement with the Social World, Chicago: Markham.
Handwerker, L. (1994), “Medical Risk: Implicating Poor Pregnant Women,” Social Science and Medicine, 38:5, 665-675.
Hedges, S. (1985), “Group Interviewing,” in R. Walker, ed., Applied Qualitative Research, Brookfield, Vermont:: Gower, 239-269.
Hernstein, R.J. and C. Murray (1994), The Bell Curve: Intelligence and Class Structure in American Life, New York: Free Press.
Holtzman (1997), “The Second Conference on Genetics and Primary Care: The Role of Primary Care Providers and Medical Educators,” in Nancy Touchette et al., Toward the 21st Century: Incorporating Genetics into Primary Health Care, Plainview, New York: Cold Spring Harbor Laboratory Press.
Kevles, Daniel J. and L. Hood (1992), The Code of Codes: Scientific and Social Issues in the Human Genome Project, Cambridge, Massachusetts: Harvard University Press.
Kitcher, Philip (1996), The Lives to Come: The Genetic Revolution and Human Possibilities, New York, New York: Simon and Schuster.
Lippman-Hand, A. and F.C. Fraser (1979), “Genetic Counseling: Parents’ Response to Uncertainty,” in C.J. Epstein et al., eds., Risk, Communication, and Decision-Making in Genetic Counseling, NY: Liss, 325-339.
Los Angeles Times (1997), “Clinton Delivers Tuskegee Apology,” Friday, May 16.
Merton, R.K., M. Fiske and P. Kendall (1956), The Focused Interview: A Manual of Problems and Procedures, Glencoe, Illinois: Free Press.
Morgan, D.L. (1988), Focus Groups as Qualitative Research, Beverly Hills, CA: Sage.
Morgan, D.L. and M.T. Spanish (1984), “Focus Groups: A New Tool for Qualitative Research,” Qualitative Sociology, 7, 253-270.
Nelkin, Dorothy and M. Susan Lindee (1995), The DNA Mystique: The Gene as a Cultural Icon, New York: Freeman.
Powell, Walter W. and Jason Owen-Smith (1997), “Universities as Creators of Intellectual Property: Life Sciences Research and Economic Development,” Paper presented at the 1997 Annual Meetings of the American Sociological Association, Toronto, Canada, August 8-13.
Rothenberg, Karen, et al. (1997), “Genetic Information and the Workpalce: Legislative Approaches and Policy Challenges,” Science, the American Association for the Advancement of Science, 275: 1755-1757, March 12.
Schutz, Alfred (1970)., On Phenomenology and Social Relations: Selected Writings. Chicago: University of Chicago Press.
Wang, Vivian and Marsh, Frank H. (1992), “Ethical Principles and Cultural Integrity in Health Care Delivery: Asian Ethnocultural Perspectives in Genetic Services,” Journal of Genetic Counseling 1 (1): 81-92, 1992
Weitz, Rose (1994), “Uncertainty and the Lives of Persons with AIDS,” pp. 138-149 in Peter Conrad and Rochelle Kern (eds.), The Sociology of Health and Illness: Critical Perspectives (New York: St. Martin’s Press), 1994.
Wertz, Dorothy C., et al. (1992), “Attitudes Toward the Prenatal Diagnosis of Cystic Fibrosis: Factors in Decision Making Among Affected Families,” American Journal of Human Genetics, 50:1077-1083.