JASON Evaluation Report of the Human Genome Project – Quality (cont’d)
Previous Section * Title and Table of Contents * Next Section
JSR-97-315
October 7, 1997
The MITRE Corporation
3. QUALITY
A project with the stated goal of sequencing the entire human genome must make data accuracy and data quality integral to its execution. It is clear that much of the genome will later be re-sequenced piece-by-piece. But a high-quality database can reduce the need for such resequencing, provide useful and dense markers across the genome, and enable large-scale statistical studies. A quantitative understanding of data quality across the whole genome sequence is thus almost as important as the sequence itself.
Technology for large-scale DNA sequencing is relatively new. While current sequencing tools and protocols are adequate at the lab-bench level, they are not yet entirely robust. For generic DNA sequence, the mainstream techniques are straightforward and can be carried out with low error rates. However problems and errors occur more frequently when sequencing particular portions of the genome or particular sequence patterns, and resolving them requires expert intervention. Phenomena such as deletions, unremoved vectors, duplicate reads, and chimeras are often the consequence of biological processes, and as such are difficult or impossible to eliminate entirely. Base-call accuracy tends to degrade toward the end of long sequence reads. Assembly of complete genomic sequences remains a challenge, and gaps are sometimes difficult to fill. In this situation, quality assurance and quality control (QA/QC) are essential. In particular it is crucial to understand quantitatively the accuracy of information going into the genome data base. The present section of this report discusses the coupled issues of quality assurance, quality control, and information about data quality, as they impact the Project, as well as other national and international sequencing efforts.
The following three steps provide a useful framework for analyzing and addressing QA/QC issues for the Project (indeed, for any large-scale sequencing effort):
- Quantify the quality requirements of present and future uses of genomic information
- Develop assays that can accurately and efficiently measure sequence quality
- Take steps to ensure that present and evolving sequencing methods and data meet the prescribed level of quality.
The following subsections consider each of these issues in turn. We then follow with some summary recommendations on QA and QC. Following the conclusion of our study, we became aware of a report of an NHGRI Workshop on DNA Sequence Validation held in April, 1996 [http://www.genome.gov/10001513] that independently examined some of the same issues and came to some similar conclusions.
3.1 Quality requirements
Our briefers reflected a wide range of opinions on the magnitude of the required error rates for sequence data. This has clearly been a controversial issue and, at times, it has been used as a surrogate for other inter-Center disputes. We believe that the debate on error rates should focus on what level of accuracy is needed for each specific scientific objective or end-use to which the genome data will be put. The necessity of “finishing” the sequence without gaps should be subject to the same considerations. In the present section, we stress the need for developing quantitative accuracy requirements.
3.1.1 The diversity of quality requirements
Genomic data will be (indeed, are being) put to a variety of uses and it is evident that the quality of sequence required varies widely among the possible applications. If we quantify accuracy requirements by the single-base error, ε, then we can give some representative estimates:
| Application | Error requirement |
|---|---|
| Assemble long contigs | ε ~10-1 |
| Identify a 20-mer sequence | ε ~10-1 |
| Gene finding | ε ~ 10-2 |
| Construct a pair of 20-mer STS primers | ε =2.5×10-4 (99% confidence)ε =2.5×10-3 (90% confidence) |
| Polymorphism | ε ~2.5×10-5 (coding regions)ε ~10-4 (non-coding regions) |
| Studies of genomic evolution, statistics | ??? |
Although these are only rough order-of-magnitude estimates; we justify each as follows.
- The surprisingly low accuracy we estimate to be required to assemble long contigs and to identify the presence of a precisely known 20-mer in a sequence is discussed in the following subsection for the ideal case of no repeats
- Our estimate for the gene finding requirement is based on the observation that pharmaceutical companies engaged in this activity seem satisfied with short sequences (400 bases) at this level of accuracy.
- The required accuracy to construct a pair of 20-mer STS primers is based on straightforward probabilistic calculations.
- The polymorphism entry simply repeats the common statement that accuracy 10 times better than the observed polymorphism rate is sufficient.
- The requirements for evolutionary or statistical studies of the genome have not been quantified
More precise estimates for each of these uses (and others) can surely be generated by researchers expert in each of the various applications. Beyond qualitative judgment, one useful technique would be to run each of the applications with pseudodata in which a test sequence is corrupted by artificially generated errors. Variation of the efficacy of each application with the error level would determine its error requirement and robustness. Such exercises, carried out in software, cost little, yet would go a long way toward setting justifiable quality goals. We recommend that the DOE encourage the genomics community to organize such exercises.
With this kind of data in hand, one could establish global quality requirements for the final sequence (perhaps different for coding and non-coding regions). It is likely that arbitrarily high accuracy could be achieved by expending enough effort: multiple sequencing with alternative technologies could guarantee high accuracy, albeit at unacceptable cost. In the real world, accuracy requirements must be balanced between what the users need, the cost, and the capability of the sequencing technology to deliver a given level of accuracy. Establishing this balance requires an open dialog among the sequence producers, sequence users, and the funding agencies, informed by quantitative analyses.
3.1.2 Accuracy required for assembly
A probabilistic analysis of the assembly problem shows that (in an ideal case) assembly requires relatively little accuracy from the raw sequence data. These data are the sequences of base calls derived from the individual reads. An accuracy as low as 0.9 (per base call) is sufficient to ensure reliable assembly. A high degree of coverage is required, however, to have any chance of assembling the entire clone without gaps. The following analysis justifies these statements, albeit in the absence of repeats, which are likely to complicate the situation considerably.
We first consider the problem of assembling k fragments of length L with left endpoints uniformly distributed over a clone of length M. Requiring overlaps above a given threshold does not really complicate the gap problem. The point is that a tiling of the sequence of length M with fragments of length L overlapping with subsegments of length at least x is ensured by a tiling with no gaps with fragments of length L-x.
We can compute an approximate lower bound for the probability of success as follows. The probability that for a given region of length L*, some fragment has its left endpoint somewhere in the given region is
1 – (1 – L*/M)k
where k is the number of fragments considered.
We now suppose that the clone length is 30,000 and that the fragments have length 1300. The probability that with 450 fragments there exists a sequence of 150 distinct fragments starting at the left end of the clone such that each successive fragment starts in the left-justified 1200-length subfragment of the previous fragment (thereby ensuring overlaps of 100) is at least
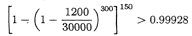 ,
,
which is conservative since the inner exponent is really varying from 449 to 300.
Randomly selecting such a walk across the clone, the probability that the walk reaches the other end of the clone is greater than
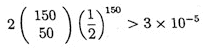 .
.
This conservatively estimates the probability that at least 50 of the successive overlaps begin in the right-justified half of the 1200 length region of the previous fragment (and so extend the walk by at least 600 bases). Thus the probability that the selected walk covers the clone is greater than 0.999.
Sequencing the fragments from both ends yields the sequence, assuming read lengths of 650. The advantage of longer reads is that longer fragments can be used and hence for a desired probability for coverage, fewer fragments can be used. A distinct possibility is that merely improving the percentage of long reads has a significant effect.
We emphasize that these are simply lower bounds which are rather conservative, computed for this idealized case.
We next consider the probability that a complete tiling can be constructed and correctly assembled given a specific error rate in the base calls. Suppose that G is a sequence of bases of length x, G* is a probabilistic garbling of G with an error rate 1-E and that R is a random sequence of length x. For each m<x, the probability that G* and G disagree in at most m places is
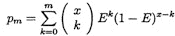 ,
,
while the probability that R and G disagree in at most m places is
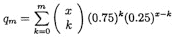 ,
,
which is dominated by the last term for the relevant values of x and m.
We examine the case when x=100 and E=0.1. In the assembly problem, pm should be calculated with a smaller error rate since one is considering matches between two garbled sequences. For an error rate of E=0.1, the effective error rate is approximately 0.186. Typical values for varying choices of m are
p39=0.9999996; p40=0.99999987; p41=0.99999995.
The corresponding values for qm are
q39=2.87 x 10-14; q40=1.33 x 10-13; q41=5.90 x 10-13.
At each stage of the construction of the walk and with a threshold of m, the probability that there is an assembly error which passes the threshold requirement is at most
1 – (1 – qm)1200×450.
The probability that a correct fragment will pass, correctly placed, is at least pm (in the worst case of there only being one such fragment). Thus, if there is a walk across the clone, the probability of constructing a valid walk across the clone is at least
Pm = (1 – qm)1200x450x150 x pm150.
With values as above, we have
P39=0.99993; P40=0.99997; P41=0.99996.
With a threshold of 40 the probability of constructing a correct walk across the clone is essentially the same (0.999) as the probability that there exists such a walk across the clone.
The analysis here makes several (important) simplifying assumptions. For example, it assumes that the fragments are uniformly distributed across the clone and that the clone itself is a random sequence of base pairs. While in some regions of the genome the latter may be a good assumption, there are certainly areas where it is not. More importantly, even somewhat limited partial repeats within the clone will have a possibly significant impact on the analysis. This can be explored experimentally via computer simulations using known stretches of the sequence (Section 3.3.1).
Further, with fragments produced using sets of restriction enzymes, the fragments may well not be uniformly distributed and we only considered pointwise garbling (not insertions or deletions). However the intent of this analysis is simply to illustrate the relative importance of base-calling accuracy and coverage (number of fragments) in the sequencing process.
Another important point is that attention should be paid to examining the relative merits of:
- Having the sequence of the genome at relatively low accuracy, together with a library of fragments mapped to the sequence;
- Having the sequence of the genome at high accuracy.
There are sequencing strategies in which the order of the fragments is essentially known in advance. The assembly of such a library of fragments is easier (significantly easier for the idealized random genome). It is possible that for sequencing certain regions of the genome these approaches, coupled to accepting higher error rates in the reads, are superior.
A final point concerning accuracy is the placement of known sequences against the garbled genome sequence. Suppose that, as above, the garble rate is 0.1; i.e., the accuracy is 0.9. Then given a sequence of length 50 from the true sequence, the probability that the sequence is correctly, and uniquely, placed is 0.999 using a threshold of 12 errors. Again, the assumptions are that the genome sequence is random or at least that the given segment is from a portion of the genome which is random. However if a significant fraction of the genome is random then (with high probability) false placements will only happen in the remaining fraction of the genome. This could be used to produce interesting kinds of maps, using a small library of target fragments. Again, some simulations can easily test these various points against known sequence data and allowing errors of insertion and deletion.
3.2 Verification protocols
Since the “proof of the pudding” lies in the actual accuracy of the output, absolute accuracy can be determined only by physical testing of the sequence output. That is, given the putative sequence of base pairs for a certain contig (which we term the “software sequence”), independent protocols should be established to verify this software sequence relative to the physical contig. Such “verification” is a different task from de novo sequencing itself, and should be accomplished by means as independent as possible from those employed to obtain the initial sequence.
An ideal verification method would be:
- Sequence blind: requires no a priori knowledge of the sequence
- Sequence independent: efficacy independent of the sequence being verified
- Reliable: a high probability of detecting errors, with low probability of false alarms
- Economical: cost (labor, materials, time) a small fraction of the cost of sequencing
- Capable: long sequences easily verified
- Specific: provides further information about the errors beyond “Right or Wrong”
One obvious strategy is to resequence the DNA by a method different than that used by the original researcher. Unfortunately, this fails on the grounds of economy and the fact that today there is really only one large-scale sequencing technique.
In this section, we describe two possible verification protocols, and close with a discussion of the implementation of any protocol.
3.2.1 Restriction enzyme verification of sequence accuracy
We propose Multiple Complete Digestions (MCD) as a verification protocol satisfying most of the criteria above. It will allow statements like “With 90% probability, this sequence is accurate at the 10-3 level” or, more generally, “With confidence C, the sequence is accurate at the ε level.” It may also be used to localize and characterize errors in the sequence.
MCD has been developed and used as a method for generating high-quality physical maps preparatory to sequencing [G. K.-S. Wong et al., PNAS 94, 5225-5230, 1997]. Here, we quantify the ability of this technique to provide probabilistic sequence verification.
The basic idea is that the putative sequence unambiguously predicts the fragment lengths resulting from digestion by any particular endonuclease, so that verification of the fragment lengths is a necessary (but not sufficient) check on the sequence. Multiple independent digestions then provide progressively more stringent tests. Of course, if the putative sequence has been generated by MCD with one set of enzymes, a completely different set must be used for verification.
Let us assume that ε is the single-base error rate, that only single-base substitutions or deletions can occur, and that we are using restriction enzymes specific to a b-base pattern (most commonly, b = 6 for the enzymes used in sequencing, although enzymes with b = 4, 5, 7, and 8 are also known).
A digestion will give an error (i.e., fragments of unexpected length) when an error has destroyed a restriction site or created a new one from a “near-site” of b-bases whose sequence differs from the target sequence by one base (we ignore the probability of two or more errors occurring simultaneously within a restriction site or near-site). Then the probability of any one restriction site being destroyed is bε (since the error can occur in any one of the b positions), while the probability of a near-site being converted is ε/3 (since only one of the three error possibilities for the “wrong base” leads to a true site).
Then the expected number of errors in a sequence containing S sites and N near sites is
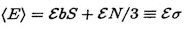
where 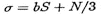 is the effective number of sites.
is the effective number of sites.
3.2.1.1 Probabilistic estimate
Let us now consider a sequence of length L bases. Assuming that bases occur at random, we expect S=L/4b sites for a single restriction enzyme and N=3bL/4b near sites, since there are 3 ways each of the b bases at a site can differ from the target pattern. Hence, for D different digestions, we expect
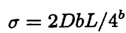 .
.
Since the number of fragments expected if there are no errors is S=L/4b and a convenient number of fragments to separate is S=10, taking b=6 implies a sequence length of L=40 kb (the size of cosmid clones) and σ= 120D = 600 if D = 5.
3.2.1.2 Real DNA
The probabilistic estimate of σ above assumed that all b-mers were equally likely, or more precisely, that the recognized b-mers were uniformly distributed. However, there is no need to make that assumption when DNA is presented for checking. Instead one can scan the proposed sequence and count the number of sites where errors could make a difference in how the sequence is cleaved. The calculation mimics exactly the random model above: each recognized site contributes 1 to σ and each near site contributes 1/3. The total for the sequence is then the contribution of that endonuclease to σ.
The table below shows the results of this counting for D=5 restriction enzymes for three pieces of human sequence from the Whitehead Center: L10 of length 48 kb, L8 of length 47 kb, and L43 of length 44 kb. (The first two are on 9q34, while the third is on the Y chromosome). Also considered is a completely random sequence of 40 kb.
| Site \ Fragment | L10 (48 kb) |
L8 (47 kb) |
L43 (44 kb) |
Random (40 kb) |
|---|---|---|---|---|
| GGATCC (BamI) |
126 | 117 | 112 | 137 |
| GATATC (EcoRV) |
49 | 40 | 105 | 94 |
| AAGCTT (HindIII) |
66 | 112 | 134 | 121 |
| TCTAGA (BglII) |
84 | 79 | 190 | 145 |
| TGGCCA (MscI) |
295 | 377 | 109 | 122 |
| σ | 620 | 725 | 650 | 619 |
These results agree with the probabilistic estimate of σ~600 for D=5 and L~40 kb. However, while the probabilistic model is true on average, it is not true in detail and some restriction enzymes give more meaningful tests of a given sequence than others (i.e., contribute more to σ). For example, digestion of L10 with EcoRV does not add very much information, while digestion with MscI does. Hence, for a given DNA sequence, it is possible to choose the most meaningful set of restriction enzymes to be used in the test.
3.2.1.3 Judging the results
When a particular sequence is digested with a particular set of enzymes, the number of errors actually observed will be given by a Poisson distribution, in which the probability of observing E errors is
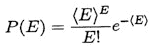 .
.
What can be learned from a MCD test that shows E errors? Let us assume that the tests are arranged so that σ=700, that ε=10-3 the quality goal, and that we declare that any sequence showing E<2 errors in an MCD test is “good.” In that case, there is a false alarm probability of PFA=0.16 that an ε=.001 sequence will be rejected, and will have to be redone. However, if the sequence has ε=0.01, there is only a PA=0.007 probability that it will be accepted. Hence, this simple operational definition (at most one error) implies only slightly more work in resequencing, but gives high confidence (>99%) in a sequence accuracy at the level of ε=0.01 and 90% confidence in the sequence at the ε~0.005 level. The implications of other choices for the maximum acceptable number of errors or for different values of <E> follow straightforwardly from the properties of the Poisson distribution; some representative values for σ=700 are given in the table below.
| E<1 | E<2 | E<3 | E<4 | |
|---|---|---|---|---|
| PFA(ε=0.001) | 0.50 | 0.16 | 0.035 | 0.006 |
| PA(ε=0.01) | 0.0009 | 0.007 | 0.03 | 0.08 |
| ε(PA=0.1) | 0.003 | 0.005 | 0.008 | 0.010 |
Note that the estimates above assume both perfect enzyme specificity; and sufficient fragment length resolution (1% seems to be achievable in practice, but one can imagine site or near-site configurations where this would not be good enough, so that a different set of restriction enzymes might have to be used). The extent to which these assumptions hinder MCD verification, as well as the ability of the method to constraint sequence to ε<10-4, can best be investigated by trials in the laboratory.
3.2.2 Hybridization arrays for sequence verification
As we have discussed in Section 2.3.3, the combinatorics make de novo sequencing a formidable challenge for present-day hybridization arrays. However, beyond the differential sequencing applications we have discussed, one potentially important application of hybridization arrays is to the problem of sequence quality control and verification, particularly since it is extremely important to employ means independent of those used to derive the putative sequence of a particular contig.
Hybridization arrays could provide a method for sequence verification independent of the present Sanger sequencing. The strategy would be to construct a Format 2 array based upon the candidate sequence for the contig. This array would then be challenged by the physical contig, with the goal being to detect differences between the “software” sequence as determined by a previous sequencing effort and the “hardware” sequence of the contig itself. For this protocol the “software” sequence would be represented by the oligomer probes of the array. Since the objective is to detect differences between two very similar sequences, the requirements on the number of distinct probes and hence on the size of the array are greatly relaxed as compared to the previous discussion of de novo sequencing. More explicitly, to scan a target contig of length N bases for single-base mismatches relative to a “known” (candidate) sequence, an array of 4N probes is required, which would increase to 5N if single site deletions were included. The array might include as well sets of probes designed to interrogate specific “problem” sections of the target. For N~40 kb, the required number of probes is then of order 2×105, which is within the domain of current commercially capability.
Note that relative to the proposal in Section 3.3.2 to establish “gold standards” of DNA sequence, this strategy could also play an important role in helping to verify independently the standards themselves.
A case study relevant to the objective of sequence verification and error detection by hybridization is the work of M. Chee et al. [op cit.], for which an array with 135,000 probes was designed based upon the complete (known) 16.6 kb sequence of human mitochondrial DNA. As illustrated in Figure 2, this work detected sequence polymorphisms with single-base resolution, with 15-mer probes. Note that the total number of probes (135,000) is considerably smaller than the total possible set for a 15-mer (415 ~ 109), allowing considerable flexibility in the design of the probes. In terms of an overall figure of merit for accuracy, the simplest possible procedure was employed whereby a scan to detect the highest fluorescent intensity from among the four possible base substitutions was made and led to 99% of the target sequence being read correctly. While this accuracy is not overwhelmingly, considerable improvement could presumably be achieved by incorporating more sophisticated analysis algorithms which take into account the overall pattern of mismatches, such as the were in fact employed by Chee et al. in their studies of polymorphisms for mitochondrial DNA from various populations. Of course since mDNA is eubacterial in character, many of the more challenging sequence pathologies are absent relative to eukaryotic DNA. Still, Chee et al. provides a useful benchmark against which to assess the potential of hybridization arrays for sequence verification.
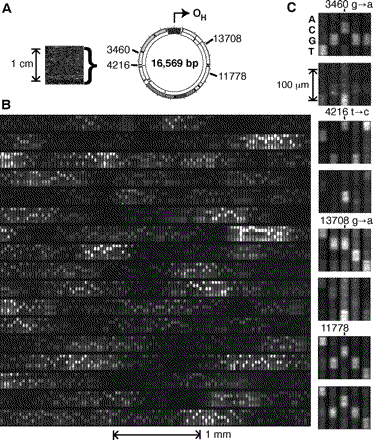
Figure 2: Human mitochondrial genome on a chip. (A) An image of the hybridized array with 135,000 probes designed to interrogate the 16.6 kb mitochondrial target RNA. (B) A magnified portion of the array. (C) Illustration of the ability to detect single base-pair differences. [from M. Chee et al., op cit.]
Perhaps the most important motivation for suggesting this strategy for verification is that the “mistakes” associated with sequence determination from target-probe interactions in a massively parallel fashion may well be sufficiently different from those arising from the gel-based procedures so as to give an independent standard for accuracy. Of course there are a host of issues to be explored related to the particular kinds of errors made by hybridization arrays (including the fidelity with which the original array is produced, hybridization equivalents, etc.). For the purpose at hand, attention should be focused on those components that most directly impact the accuracy of the comparison.
Particular suggestions in this regard relate to the readout and image processing for the array, tasks which are often accomplished site by site via scanning confocal microscopy. It would seem that alternate readout strategies should be explored, including (perhaps image-intensified) CCDs. Since the target sequence is known with small uncertainty as are the set of errors associated with single-base substitutions and deletions as well as with other “typical” errors in sequence reconstruction, image templates could be pre-computed and cross-correlated with the actual image by adapting algorithms from the image processing community to improve the accuracy with which information is extracted from the array.
The value of such a strategy for sequence verification extends beyond that of providing an independent avenue for error checking. It would also couple the traditional gel-based effort to emerging advanced technologies, with potential benefit to both. Moreover, it could be part of a broader attempt to define a longer-term future for the sequencing centers as new technologies come on line to supplant gel-based sequencing and as emphasis shifts from de novo sequencing to comparative studies such as related to polymorphisms.
3.2.3 Implementation of verification protocols
Any verification protocol must require significantly less effort that resequencing, and so there will be considerable latitude in its implementation. In one limit, sequencing groups might be required to perform and document verification protocols for all finished sequence that they wish to deposit in a database. Alternatively, a “verification group” could be established to perform “spot” verifications of database entries selected at random. A third possibility is to offer a “bounty” for identifying errors in a database entry.
Clearly, economic, sociological, and organizational factors must be considered in choosing among these, and other, possible implementations. We recommend that the funding agencies promote a dialog within the sequencing communities about possible verification protocols and their implementation.
3.3 Assessing and improving present techniques
Our emphasis on quantitative metrics for accuracy is made against the backdrop of inadequate understanding of the quality of the “end product” in the current Human Genome sequencing effort. While the level of competence and effort devoted to “doing the job right” in the sequencing centers is commendable, there is a clear need to implement a comprehensive program of quality assurance and quality control. Here we suggest some ways to provide more quantitative measures of the errors in the end product, and to understand how the various steps in sequencing contribute to the overall error budget.
Quality assurance and quality control (QA/QC) are of sufficient importance to be made integral requirements in the Project. Each sequencing center should invest a fraction of its own budget to characterize and understand its particular accuracy and error rates. This should be part of a continuing effort, rather than a one-time event. Quality control within the Centers should not be externally micro-managed, but each Center should be required to develop its own credible plan for QA/QC.
We further urge that the effort to develop new QA/QC technology be tightly coupled to the sequencing centers. In particular, new technologies such as large scale hybridization arrays or single-molecule sequencing are not currently competitive with gel-based electrophoresis for high throughput sequencing and long base reads, but they could be exploited in the short term for “niche” applications such as sequence verification for QA/QC. In the longer term, the Centers must integrate new technical advances into their operations, and the avenue of QA/QC is an important mechanism to help to accomplish this goal. From a longer-term perspective it seems clear that after the human genome has been sequenced once, emphasis will shift toward differential sequencing relevant to the study of polymorphism and homologies, and to the genetic origins of disease. QA/QC can thus be viewed as part of a broader effort to define a long-term future for the sequencing Centers, with technological leadership at the forefront as a prime component.
3.3.1 A systems approach is required
As we have noted, the issues of accuracy and error rates in reconstructed genomic information are crucial to the value of the”end-product” of the Human Genome Project, yet requirements for accuracy are complex and detail-dependent. DOE should support a portfolio of research in genome quality assurance and quality control issues.
One of the elements of this research should be computer simulation of the process of sequencing, assembly, and finishing. We believe that research into the origin and propagation of errors, through the entire system, are fully warranted. We see two useful outputs from such studies: 1) more reliable descriptions of expected error rates in final sequence data, as a companion to database entries, and 2)”error budgets” to be assigned to different segments of mapping and sequencing processes to aid in developing the most cost-effective strategies for sequencing and other needs.
DOE should solicit and support detailed Monte Carlo computer simulation of the complete mapping and sequencing processes. The basic computing methods are straight-forward: an reference segment of DNA (with all of the peculiarities of human sequence, such as repeats) is generated and subjected to models of all steps in the sequencing process; individual bases are randomly altered according to models of errors introduced at the various stages; the final, reconstructed segment or simulated database entry is compared with the input segment and errors are noted.
Results from simulations are only as good as the models used for introducing and propagating errors. For this reason, the computer models must be developed in close association with technical experts in all phases of the process being studied so that they best reflect the real world. This exercise will stimulate new experiments aimed at the validation of the error-process models, and thus will lead to increased experimental understanding of process errors as well.
Useful products of these types of simulations are”error budgets” for different steps in the measurement and analysis chain. Such budgets reflect the contributions of individual steps and their effect on the accuracy of the final result. This information can be used, for example, to establish quality criteria for the various stages of the sequencing process, so that emphasis and funds can be devoted to improving the accuracy of those steps which have the strongest influence on the accuracy of the final sequence product.
Error budgets will depend on the final accuracy required for a specific use of the end-product, which is analyzed sequence information. By comparing specific end-product needs for accuracy and quantity of information with error budgets and costs of individual steps in the overall process from DNA to database, it should be possible to perform cost/benefit analyses for developing optimum sequencing strategies.
3.3.2 Gold standards for measuring sequence accuracy
DOE should take the lead in developing “gold standards” for human DNA sequence. Standard DNA sequences could be used by the whole sequencing community for assessing the quality of the sequence output and sequencing protocol through “blind” experiments within the various centers. These gold standards should be designed to highlight quality assessment in “hard” DNA-sequencing regions and in potential problem areas, as well as in “ordinary” coding regions. They would consist of cloned DNA molecules of two types:
- a cosmid vector containing an insert of ~40 kb of human DNA that has been sequenced with high accuracy and assembled without any unresolved discrepancies;
- a phagemid vector containing an insert of ~1 kb of synthetic DNA including both human-derived sequences and contrived sequences that are known to cause common artifacts in DNA sequence acquisition.
The standard cosmid will have been transduced and propagated in bacterial cells, then stored as individual aliquots kept at -70 °C. Upon request, one or more of these aliquots would be made available to a sequencing group. All of the subsequent steps, including further propagation of the cosmid, restriction mapping, subcloning, sequencing, assembly, and finishing would be carried out by the sequencing group. Performance could be assessed based on a variety of indices such as PHRED and PHRAP scores, number of sequencing errors relative to the known standard, type of sequencing errors, time required to complete the sequencing, and cost of sequencing. The cosmid standard might also be used to compare alternative sequencing protocols within a sequencing center or to conduct pilot studies involving new instrumentation.
The standard phagemid will have been produced in large quantity, purified, and stored as individual aliquots kept at -70 °C. After thawing, the DNA will be ready for sequencing, employing “universal” primers that either accompany the template DNA or are provided by the sequencing group. The purpose of this standard is to assess the quality of DNA sequencing itself, based on indices such as PHRED score, read length, and the number and type of sequencing errors relative to the known standard. The target sequence will have been designed to elicit common sequencing artifacts, such as weak bands, strong bands, band compressions, and polymerase pauses.
Although the cosmid standard is expected to have greater utility, the phagemid standard will be used to control for variables pertaining to DNA sequencing itself within the overall work-up of the cosmid DNA. It is likely that the sequencing groups will be on their “best behavior” when processing a gold standard, resulting in enhanced performance compared to what might be typical. This cannot be avoided without resorting to cumbersome procedures such as surprise examinations or blinded samples. Thus it will be important to examine not only the output of the sequencing procedures, but also the process by which the data is obtained. The extent to which it is possible to operate in a “best behavior” mode will itself be instructive in assessing DNA sequencing performance. At the very least, such trials will establish a lower limit to the error rate expected.
We recommend that the DOE provide funding, on a competitive basis, to one or two individual investigators who will construct and maintain the DNA standards. It might be appropriate to construct a small family of cosmid and phagemid standards that would be made available sequentially. The experience of the sequencing groups in processing these gold standards will suggest ways in which they could be improved to better assess critical aspects of the sequencing process.
3.3.3 Quality issues pertaining to sequencing templates
While most of our discussion has involved QA/QC issues in the sequencing and assembly process, it is useful to consider also quality issues in the processes used to prepare DNA for sequencing. We do so in this subsection.
There are many steps involved in construction of a human genomic DNA library and subcloning of that library into a form suitable for automated DNA sequencing. These include:
- fragmentation of chromosomal DNA by mechanical shearing or partial enzymatic digestion;
- size fractionation of the DNA fragments by gel electrophoresis or centrifugation;
- cloning of ~1 Mb fragments into high-capacity vectors, such as YACs or BACs;
- propagation of YACs or BACs within host cells;
- enzymatic digestion of YAC or BAC inserts to obtain fragments of ~40 kb;
- cloning into medium-capacity cosmid vectors;
- propagation of cosmids within bacterial cells;
- enzymatic digestion of cosmid inserts to obtain fragments of ~1 kb;
- cloning into low-capacity plasmid or phagemid vectors;
- preparation of purified plasmid or phagemid DNA.
The subsequent steps of dideoxy sequencing, base calling, assembly, and finishing are all prone to errors that can be investigated and quantified, as we have discussed in previous sections. However, each of the steps above can introduce artifacts that make sequencing more difficult.
The steps involved in the preparation of templates for sequencing are made error-tolerant by the exponential amplification that is inherent in these procedures. Errors do occur, such as empty vectors, poor transformation efficiency, insufficient vector amplification, and inadequate purity of the template DNA. These problems usually result in clones that drop out of the process. Provided that there is redundant coverage of the DNA among the successful clones, the failed clones can essentially be ignored. However three quality control issues pertaining to template preparation merit special attention:
- There may be incomplete representation of the genomic DNA at the level of the BAC/YAC, cosmid, or plasmid/phagemid libraries. This may be due to insufficient redundancy in construction of the library, but more often they are due to regions of the chromosome that are either difficult to clone or difficult to propagate within host cells. The genomics community is well aware of these problems and has taken appropriate countermeasures. Unlike the yeast genome, which has been sequenced successfully in its entirety, there may be regions within the human genome that cannot be cloned and therefore cannot be sequenced. At present the best course of action is to press ahead and deal with the problem of “unsequenceable” DNA if and when it arises.
- There may be spurious DNA sequences intermixed with the desired genomic DNA. The two most common sources of contamination are vector-derived DNA and host cell DNA. Vector sequence can be recognized easily by a suitable sequence-matching algorithm. Incredibly, there are many entries in the genomic databases today that are either partly or completely derived from vector sequence. Host cell DNA is more difficult to recognize, but these too can be identified with the complete genomic sequences of yeast and E. coli available. Although spurious sequences can be eliminated after the fact, it should be made incumbent on the sequencing centers to do this prior to database submission.
There are challenges in maintaining proper inventory control over the vast number of clones and subclones that are being generated by the Project. Current procedures at the major genome centers are adequate in this regard. A physical inventory should be maintained for all BAC/YAC and cosmid clones, but this is not critical for the plasmid/phagemid clones. An electronic inventory, with secure back-up copies, should be maintained for all clones and subclones that are generated.