JASON Evaluation Report of the Human Genome Project – Background (cont’d)
Title and Table of Contents * Next Section
JSR-97-315
October 7, 1997
The MITRE Corporation
1. BACKGROUND, CHARGE, AND RECOMMENDATIONS
1.1 Overview of the Human Genome Project
The US Human Genome Project (the “Project”) is a joint DOE/NIH effort that was formally initiated in 1990. Its stated goal is
“…to characterize all the human genetic material–the genome–by improving existing human genetic maps, constructing physical maps of entire chromosomes, and ultimately determining the complete sequence… to discover all of the more than 50,000 human genes and render them accessible for further biological study.”
The original 5-year plan was updated and modified in 1993 [F. Collins and D. Galas,”A new five-year plan for the US Human Genome Project,” Science 262, 43-46 (1993)]. The Project’s goals to be achieved by the end of FY98 that are relevant for this study are:
- To complete an STS (Sequence Tagged Site) map of the entire genome at 100 kb resolution
- To develop approaches for sequencing Mb regions
- To develop technology for high-throughput sequencing, considering the process as integral from template preparation to data analysis.
- To achieve a large-scale sequencing capacity of 50 Mb/yr and to have completed 80 Mb of human sequence
- To develop methods for identifying and locating genes
- To develop and disseminate software to archive, compare, and interpret genomic data
Congress has authorized funding through the planned completion of the Project in FY05. The funding in FY97 is $189M for the NIH activity and $78M for the DOE. Thus the total US effort is $267M this year. This amounts to more than half of the worldwide effort, with France, UK, the EU, and Japan being the other major partners.
The DOE program in FY97 included $29M for production sequencing, $15M for the startup of the Joint Genome Institute (a “factory scale” sequencing facility to be operated jointly by LLNL, LANL, and LBNL), $13M for technology development, $11M for informatics, and $3M for applications (construction of cDNA libraries, studying gene function, etc.)
1.2 Challenges for the Project
There are a number of challenges that the Project faces if it is to meet its stated goals. We briefly describe several of them in this section as a background to our charge.
1.2.1 The complexity of genomic data
One of the challenges to understanding the genome is the sheer complexity of genomic data. Not all sequence is equivalent. The 3-5% of the genome that is coding consists of triplet codons that specify amino acid sequence. The control regions are binding sites for regulatory proteins that control gene expression. The functions of the introns within a gene and the intergenic regions are largely unknown, even though they comprise the bulk of the genome. There are also special structural elements (centromeres and telomeres) that have characteristic base patterns.
Even given the sequence, the genes are not manifest. And the function and control of a particular gene (When and where is it expressed? What is the function of the protein it encodes?) generally must be determined from the biological context, information beyond the bare sequence itself.
Yet another challenge is that the genomes of any two individuals (except of identical twins) are different (at the 10-3 level in the non-coding region; 3-5 times less in the coding regions), and that the homologies between organisms are invariably less than perfect.
Many of these difficulties arise because we don’t yet understand the language of the genome. A good metaphor for the state of genetic information is “It’s like going to the opera.” That is, it’s clear something substantial is happening and oftimes it’s quite beautiful. Yet we can’t really know what’s going on because we don’t understand the language.
1.2.2 The state of technology
Another hurdle for the Project is the state of technology. The present state of the art is defined by Sanger sequencing, with fragments labeled by fluorescent dyes and separated in length by gel electrophoresis (EP). A basic deficiency of the present technology is its limited read-length capability (the number of contiguous bases that can be read); best current practice can achieve 700-800 bases, with perhaps 1000 bases being the ultimate limit. Since interesting sequence lengths are much longer than this (40 kb for a cosmid clone, 100 kb or more for a gene), the present technology requires that long lengths of DNA be fragmented into overlapping short segments (~1 kb long) that can be sequenced directly. These shorter reads must then be assembled into the final sequence. Much of the current effort at some sequence centers (up to 50%) goes into the assembly and finishing of sequence (closing gaps, untangling compressions, handling repeats, etc.). Longer read lengths would step up the pace and quality of sequencing, although the problem of compressions would still remain.
However, it is important to realize that, beyond the various genome projects, there is little pressure for longer read lengths. The 500-700 base reads allowed by the current technology are well-suited to many scientific needs (pharmaceutical searches, studies of some polymorphisms, studies of some genetic diseases). Thus, the goal of the entire sequence implies unique technology needs, for which there are limited medical or pharmaceutical needs.
Other drawbacks of the present technology include the time- and labor-intensive nature of gel preparation and running and the comparatively large sample amounts required to sequence. This latter influences the cost of reagents involved, as well as the necessity for extra PCR steps.
1.2.3 The pace of sequencing
One regularly updated “score card” of the Human Genome Project is maintained at http://weber.u.washington.edu/~roach/human_genome_progress2.html. This site regularly updates its tallies from the standard human genome databases. As of 11/20/97, there was some 80 Mb of human sequence in contigs of 10 kb or longer; this has been accumulated over the past 20 years. Although 97.3% of the genome thus remains to be sequenced, 40 Mb have been added in the past six months. Figure 1 below shows the progress in the past few years.
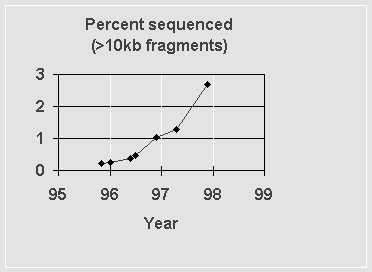 Figure 1: Fraction of the human genome in contigs longer than 10 kb that is deposited in publically accessible databases.
Figure 1: Fraction of the human genome in contigs longer than 10 kb that is deposited in publically accessible databases.
The world’s large-scale sequencing capacity is estimated to be roughly 100 Mb/yr; although not all of this resource is applied to the human genome. The Joint Genome Institute is projected to have a sequencing capacity of 57 Mb/yr in FY98, growing to 250 Mb/yr in FY01. These capacities are to be compared with the Project’s 9/98 goal of 50 Mb/yr.
It is sobering to contemplate that an average production of 400 Mb/yr is required to complete the sequence “on time” (i.e., by FY05); this corresponds to a daily generation of 50,000 samples and 15 Gbytes of raw data (if the EP traces are archived). Alternatively, an FY98 capacity of 50 Mb/yr must double every 18 months over the next seven years. These figures correspond to an ultimate scale-up of the present capacity by a factor of 30-100. Most observers believe that significant technology advances will be required to meet the FY05 goal.
The length of the known human sequences is also important. The Project’s goal is the contiguous sequence of the entire genome. The table below (taken from weber.u.washington.edu/~roach/human_genome_progress2.html) shows the number of known contiguous segments that are equal to or greater than a specified cut-off length. Note that only 1/3 of the known sequence is in lengths of 100 kb or greater, and that the longest human contig is about 1 Mb. It should also be noted that there are many known sequences of several hundred bases or less, for cDNA fragments of this size are generated at a prodigious rate by the public Merck-Washington University collaborative effort and in the pharmaceutical industry. (We heard of one company, Incyte, which produces 8 Mb of raw sequence each day, albeit in small fragments.)
| Length cutoff (kb) |
Contigs longer than cutoff |
Sequence in contigs (Mb) |
|---|---|---|
| 100 | 112 | 16.15 |
| 50 | 191 | 22.06 |
| 40 | 302 | 26.82 |
| 30 | 494 | 33.72 |
| 20 | 579 | 35.85 |
| 10 | 782 | 38.66 |
| 5 | 1227 | 41.72 |
| 1 | 5283 | 50.50 |
| 0.1 | very many |
1.2.4 The cost of sequencing
The cost of sequencing is also a major consideration. If funding continues at the present rate over the next 8 years, the US Project will spend some $2.5B. If all of this funding were devoted to production sequencing, a cost of roughly $1 per base would suffice. But only a fraction of it is.
Several cost benchmarks are available. The tenth complete microbial genome (Bacillus subtilis) has just been announced. It consists of 4000 genes in 4.2 Mb of sequence. This joint European/Japanese project cost something over $2 per base sequenced. Best practice in the Human Genome Project is currently $0.5/base, and the project goal is less than $0.10/base. Specific plans for the Joint Genome Center project an initial (FY97) cost of $0.60 per base, falling to $0.10 per base by FY01. It should be noted that there is difficulty in comparing the costs claimed across laboratories, and across the different funding systems in different nations.
1.2.5 Project coordination
The Human Genome Project presents an unprecedented set of organizational challenges for the biology community. Success will require setting objective and quantitative standards for sequencing costs (capital, labor, and operations) and sequencing output (error rate, continuity, and amount). It will also require coordinating the efforts of many laboratories of varying sizes supported by multiple funding sources in the US and abroad.
A number of diverse scientific fields have successfully adapted to a “Big Science” mode of operation (nuclear and particle physics, space and planetary science, astronomy, and oceanography being among the prominent examples). Such transitions have not been easy on the scientists involved. However, in essentially all cases the need to construct and allocate scarce facilities has been an important organizing factor. No such centripetal force is apparent (or likely) in the genomics community, although the Project is very much in need of the coordination it would produce.
1.3 Study charge
Our study was focused on three broad areas:
- Technology: Survey the state-of-the-art in sequencing. What are the alternatives beyond gel electrophoresis? What strategies should be used for inserting new technologies into production sequencing? What are the broader uses of sequencing technologies? What are the technology needs beyond those of the Human Genome Project?
- Quality Assurance and Quality Control: What are the end-to-end QA/QC issues and needs of the Human Genome Project? What levels of sequence quality are required by various users of genome data? What steps can be taken to ensure these various levels of quality?
- Informatics: Survey the current database issues, including data integrity, submission, annotation, and usability? What is the current state of algorithm development for finishing and annotating sequence?
Beyond briefings focused on these specific topics, we also heard a variety of speakers on functional genomics, in order to better get a sense of the needs, standards, and expectations of the consumers of genomic information.
Our recommendations in response to this charge are given in the following section. The balance of this report provides the necessary context and detail, dealing successively with Technology (Section 2), Quality (Section 3), and Informatics (Section 4).
1.4 Recommendations
1.4.1 General recommendations
We begin with two recommendations pertinent to many aspects of the Human Genome Project.
“Know thy system”. It is important to have a comprehensive, intimate, and detailed understanding of the sequencing process and the uses of genomic data. Gaining such understanding is quite a different exercise from sequencing itself. Answers to questions such as “What are the pacing factors in production sequencing?” (cloning? gel prep? run time?, lane count?, read length?, …) or “What is the sequence error budget?” or “What quality of sequence is required?” are essential to optimizing the Project’s utility and use of resources.
Couple users/providers of technology, sequence, data. The Human Genome Project involves technology development, production sequencing, and sequence utilization. Greater coupling of these three areas can only improve the Project. Technology development should be coordinated with the needs and problems of production sequencing, while sequence generation and informatics tools must address the needs of data users. Promotion of such coupling is an important role for the funding agencies.
1.4.2 Technology recommendations
Technology development should be emphasized as a DOE strength. Technology development is essential if the Human Genome Project is to meet its cost, schedule, and quality goals. DOE technology development leverages traditional and extensive Department expertise in the physical sciences, engineering, and the life sciences. These are, in many ways, complementary to NIH strengths and interests. If the DOE does not continue to play a leading role in technology development for high-throughput, high-capacity sequencing, it is not clear to us who will.
Continue work to improve present technologies. Although a number of advanced sequencing technologies look promising, none are sufficiently mature to be candidates for the near-term major scale-up needed. Thus, it is important to support research aimed at improving the present Sanger/EP effort. There are clear hardware and software opportunities for improving gel reading capabilities; formation of an ABI user group might accelerate the realization and dissemination of these improvements. There are also software opportunities to improve the crucial assembly and finishing processes, for example by developing a common set of finishing rules, as discussed in Section 2.1.2.
Enhance long-term technology research. The present sequencing technology leaves much to be desired and must be supplanted in the long term if the potential for genomic science is to be fully realized. Promising directions at present for advanced technology development include single-molecule sequencing, mass spectrometric methods, hybridization arrays, and micro-fluidic capabilities. The total FY97 funding for advanced technology (i.e., non-EP based) was only $1.7M of the roughly $13M total technology funding in the $78M DOE Human Genome Project; it should be increased by approximately 50%.
Retain technology flexibility in production sequencing facilities. Because sequencing technology should (and is likely to) evolve rapidly (ideally, both evolutionary and revolutionary changes will occur before FY05) it is important to retain the flexibility to insert new technologies into the large-scale sequencing operations now being created (e.g., the Joint Genome Institute). The decisions of when to freeze technology and how much upgrade flexibility to retain are faced in most large scientific projects (e.g., spacecraft or accelerators) and, unfortunately we have no magic prescription for dealing with them. However, the common sense steps of building in modularity and of thoroughly and frequently scanning the technology horizon are well worth remembering.
1.4.3 Quality recommendations
Work to make quality considerations an integral part of the Project. Quality issues must be brought to the fore in the sequencing community, since measures of sequence quality will greatly enhance the utility of the Human Genome Project’s “product.” Among the top-level steps that should be taken are allocating resources specifically for quality issues and establishing a separate QA/QC research program (perhaps a group at each sequencing center).
Quantify QA/QC issues. Promote research aimed at quantifying (through simulation and other methods) the accuracy required by various end uses of genomic data. Further, since accuracy is a full-systems issue, there is the need for a comprehensive, end-to-end analysis of the error budget and error propagation in the sequencing process, from clone library development through sequencing to databases and analysis software. “You can’t discuss it if you can’t quantify it.”
Develop and implement QA/QC protocols. Develop, distribute, and use “gold standard” DNA as tests of sequencing centers (Section 3.3.2). Support research aimed at developing, validating, and implementing useful verification protocols, along the lines discussed in Section 3.2. Make quality assessments an integral part of all database sequence. A good start would be to require that all database entries include quality scores for each base call. Existing sequencing software tools such as PHRED, PHRAP, and CONSED produce figures of merit for base calls and DNA assembly. While there is room for innovative research aimed at improving the basis for these figures of merit, the existing confidence indicators are nevertheless quite informative and should be made available to users of sequence data.
1.4.4 Informatics recommendations
Listen to the customers. Adhere to a “bottom-up”, “customer” approach to informatics efforts supported by DOE. Encourage forums, including close collaborative programs, between the users and providers of DOE-supported informatics tools, with the purposes of determining what tools are needed and of training researchers in the use of new tools and methods. Further, critically evaluate DOE-supported informatics centers with regards to the actual use of their information and services by the community.
Encourage standardization. Encourage the standardization of data formats, software components and nomenclature across the community. Invest in translators if multiple formats exist. Modularize the functions of data archiving, data retrieval, and data manipulation. Distribute the effort for development across several groups. Standardization of data formats allows more than one group to work in each area.
Maintain flexibility. Do not demand that “one-size” (in databases) fits all. Make it easy to perform the most common operations and queries, but do not make it impossible for the expert user to execute complicated operations on the data. The community should be supporting several database efforts and promoting standardized interfaces and tools among those efforts.